UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
(Mark One)
|
| |
ý | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE FISCAL YEAR ENDED December 31, 2015
OR
|
| |
¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
FOR THE TRANSITION PERIOD FROM TO
Commission File Number 001-36365
SCYNEXIS, Inc.
(Exact name of registrant as specified in its charter)
|
| | |
Delaware | | 56-2181648 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification No.) |
|
| | |
101 Hudson Street Suite 3610 Jersey City, NJ | | 07302 - 6548 |
(Address of principal executive offices) | | (Zip Code) |
(201) 884-5485
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
| | |
Title of Each Class | | Name of Each Exchange on Which Registered |
Common Stock, par value $0.001 per share | | The NASDAQ Stock Market LLC |
| | |
Securities registered pursuant to section 12(g) of the Act: None |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ¨ No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes ý No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
|
| | | | | | |
Large accelerated filer | | ¨ | | Accelerated filer | | ý |
| | | |
Non-accelerated filer | | ¨ (Do not check if a smaller reporting company) | | Smaller reporting company | | ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No ý
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant based upon the closing price of its Common Stock on the Nasdaq Global Market on June 30, 2015 was $95,905,897. Excludes 2,993,036 shares of the registrant's Common Stock held by executive officers, directors and other affiliates of registrant's Common Stock outstanding at June 30, 2015. Exclusion of such shares should not be construed to indicate that any such person possesses the power, direct or indirect, to direct or cause the direction of the management or policies of the registrant or that such person is controlled by or under common control with the registrant.
As of March 1, 2016, there were 13,905,599 shares of the registrant’s Common Stock outstanding.
Documents Incorporated by Reference
Portions of the registrant’s proxy statement to be filed with the Securities and Exchange Commission pursuant to Regulation 14A in connection with the registrant’s 2016 Annual Meeting of Stockholders, which will be filed subsequent to the date hereof, are incorporated by reference into Part III of this Form 10-K. Such proxy statement will be filed with the Securities and Exchange Commission not later than 120 days following the end of the registrant’s fiscal year ended December 31, 2015.
SCYNEXIS, INC.
ANNUAL REPORT ON FORM 10-K
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2015
TABLE OF CONTENTS
|
| | |
| | |
Item 1. | | |
Item 1A. | | |
Item 1B. | | |
Item 2. | | |
Item 3. | | |
Item 4. | | |
| | |
| | |
Item 5. | | |
Item 6. | | |
Item 7. | | |
Item 7A. | | |
Item 8. | | |
Item 9. | | |
Item 9A. | | |
Item 9B. | | |
| | |
| | |
Item 10. | | |
Item 11. | | |
Item 12. | | |
Item 13. | | |
Item 14. | | |
| | |
| | |
Item 15. | | |
| | |
| | |
| | |
| | |
| | |
| | |
| | |
PART I
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, which are subject to the “safe harbor” created by those sections. Forward-looking statements are based on our management’s beliefs and assumptions and on information currently available to our management. In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “project,” “predict,” “potential” and similar expressions intended to identify forward-looking statements. These statements involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance, time frames or achievements to be materially different from any future results, performance, time frames or achievements expressed or implied by the forward-looking statements. We discuss many of these risks, uncertainties and other factors in this Annual Report on Form 10-K in greater detail under the heading “Risk Factors.” Given these risks, uncertainties and other factors, you should not place undue reliance on these forward-looking statements. Also, these forward-looking statements represent our estimates and assumptions only as of the date of this filing. You should read this Annual Report on Form 10-K completely and with the understanding that our actual future results may be materially different from what we expect. We hereby qualify our forward-looking statements by our cautionary statements. Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
Overview
SCYNEXIS is a pharmaceutical company committed to the development and commercialization of novel anti-infectives to address significant unmet therapeutic needs. We are developing our lead product candidate, SCY-078, as a novel oral and intravenous (IV) drug for the treatment of several fungal infections, including serious and life-threatening invasive fungal infections. SCY-078 is a novel and structurally distinct glucan synthase inhibitor that has been shown to be effective in vitro and in vivo in animal studies against a broad range of Candida and Aspergillus species, including drug-resistant strains, and we are continuing to conduct additional in vitro and in vivo studies to further characterize the spectrum of activity of SCY-078. Candida and Aspergillus species are the fungi responsible for approximately 85% of all invasive fungal infections in the United States and Europe. We have completed multiple Phase 1 studies with the oral formulation of SCY-078 and we are currently conducting our first Phase 1 study with the IV formulation of SCY-078. We are also conducting two Phase 2 studies with the oral formulation of SCY-078:
| |
• | the first study is evaluating the safety, tolerability, and pharmacokinetics of SCY-078 as oral step-down treatment in patients initially treated with IV echinocandin therapy for invasive Candida infections and; |
| |
• | the second study is evaluating the safety and efficacy of orally administered SCY-078 for the treatment of vulvovaginal candidiasis (VVC). |
SCY-078 holds both Fast Track and Qualified Infections Disease Product (QIDP) designations for the IV and oral formulations for the indications of invasive candidiasis (including candidemia) and invasive aspergillosis. We expect to complete and report top line data associated with our two Phase 2 studies, our Phase 1 study and our additional in vitro and in vivo studies by the end of the second quarter of 2016.
As a spinout from Aventis S.A., or Aventis in 2000, we began as a chemistry and animal health services company, providing contract research services to third parties. Through the provision of these contract research and development services, we built significant expertise in parasitic infections and drug discovery, including expanded animal health capabilities. This contract research and development services business, which we refer to as our “Services Business,” generated substantially all of our revenue until we completed the sale of the Services Business to Accuratus Lab Services, Inc. in July 2015, as described further in the “Recent Developments” section of Item 7 of this Annual Report. Since our formation, in addition to SCY-078 and related antifungal compounds, we have discovered a number of proprietary compounds, including those within our cyclophilin inhibitor platform. We are currently focusing our resources on the development of SCY-078. In the future, we may develop other assets within our proprietary portfolio of antifungal or cyclophilin inhibitor compounds either in-house or through collaborations with strategic development partners. Additionally, we may assess external opportunities to expand our clinical pipeline.
Market Opportunity
We estimate that, each year, there are over 600,000 cases of invasive fungal infections caused by various species of Candida and Aspergillus, the two most common invasive fungal pathogens, globally. The estimated incidence in the U.S. for these conditions is approximately 98,000 and 46,000 for invasive candidiasis and invasive aspergillosis, respectively. The rapid progression of disease and high mortality rates associated with documented invasive fungal infections often result in antifungal therapy being administered in suspected (unconfirmed) cases or as a preventative measure in patients at high risk. For example, we estimate the total number of patients treated for suspected invasive Candida infections to be approximately three to four times the number of confirmed cases. Also, the increasingly widespread use of immune suppressive drugs as cancer chemotherapy or for organ transplantation or treatment of autoimmune disease has resulted in an increasing population of patients at risk for invasive fungal infections. Furthermore, the limited number of antifungal drug classes, consisting of azoles, echinocandins and polyenes, and their widespread use, has led to increased numbers of infections with drug-resistant strains. The Centers for Disease Control and Prevention (CDC), has listed fluconazole-resistant Candida as a serious threat requiring prompt and sustained action.
Among the antifungal classes available for the treatment of invasive fungal infections, only the azoles can be administered orally, imposing the need to use IV antifungal agents for the treatment of infections caused or suspected to be caused by azole-resistant Candida or Aspergillus strains. Current treatment guidelines in the U.S. and in Europe recommend the use of echinocandins (the only glucan synthase inhibitor currently commercially available) as the initial therapy for invasive candidiasis. The main limitation of the echinocandin class is that only IV administration is available, limiting the flexibility of stepping down to an oral therapy in the same treatment class. The ability to step down to an oral therapy is important because it allows the patient to be discharged from the hospital, as intravenous administration generally must occur in the hospital. The only option currently available to step down to an oral therapy after initial IV echinocandin are the azoles, which have a different mechanism of action from echinocandins (i.e., not glucan synthesis inhibitors), and have reported an increased frequency of resistant isolates, thus creating a gap in optimal treatment options for patients able to tolerate oral therapies.
Vulvovaginal Candidiasis, commonly known as a "yeast infection," is usually caused by Candida albicans but non-albicans species of Candida, such as Candida glabrata, have increasingly been identified as a cause of VVC. The estimated annual incidence in the U.S. for this condition is approximately six million cases and approximately 7-9% of these cases are considered recurrent and among the most difficult to treat. Current treatments for acute VVC include topical antifungals and the use of prescription oral antifungals such as fluconazole, which has a therapeutic cure rate of 55% as reported in its label. There are no products currently approved for the treatment of recurrent VVC.
SCY-078 represents a new chemical class of glucan synthase inhibitors designed to block an established target in infectious fungi. SCY-078 is being developed as oral and IV formulations and has demonstrated potent in in vitro activity against a large collection of medically relevant strains of Candida and Aspergillus, including multi-drug resistant strains that have been isolated from infected patients. We have conducted studies of SCY-078 using animal models that were used in the development of previously approved antifungal drugs, where these models were proven to be predictive of efficacy in humans. Using these well-established animal models, SCY-078 was shown to be highly active against Candida and Aspergillus species. SCY-078 blood concentrations were measured in a subset of the studies in the murine (i.e., mouse) model of candidiasis to determine levels required for efficacy. In subsequent Phase 1 studies, in healthy human volunteers receiving the oral formulation of SCY-078, the blood concentrations of SCY-078 achieved and met the levels predicted to be effective in treating invasive Candida infections in the animal models and, at these exposures, SCY-078 orally administered was well tolerated.
SCY-078 has unique attributes that define its potential to address significant unmet medical needs and market opportunities, including:
| |
• | only glucan synthase inhibitor with both oral and IV formulations in clinical development, allowing first-line treatment and oral step down in the same class; |
| |
• | distinct chemical structure from other glucan synthase inhibitors, allowing unique spectrum of activity and pharmacokinetic profile; |
| |
• | fungicidal (i.e., killing the fungi) capabilities against Candida species compared to azoles, which are fungistatic (i.e., inhibiting the growth of fungi); |
| |
• | activity against azole- and most echinocandin-resistant Candida strains; and |
| |
• | activity against azole-resistant Aspergillus strains. |
Based on SCY-078’s attributes and limitations of current approved therapies, we believe, if approved, our product can address therapeutic needs in three primary indications:
| |
• | recurrent vulvovaginal candidiasis, or recurrent VVC (commonly known as a "yeast infection") |
In the future we may also consider other indications for SCY-078, including prophylaxis and use for chronic invasive fungal infections.
If approved for the indications of invasive candidiasis and invasive aspergillosis, we intend to market SCY-078 to hospitals and major medical centers, where critical care physicians, infectious disease specialists, and physicians treating immune-compromised patients, such as oncologists and those performing solid organ transplants and stem cell transplants, are likely to be found and where invasive fungal infections are more prevalent. If approved for the indication of vulvovaginal candidiasis, we intend to market SCY-078 to physicians specializing in women's health.
Our positioning strategies are as follows:
| |
• | Fluconazole-resistant Candida. C. albicans is the predominant cause of fungal infections and represents a serious public health threat with significant medical and economic importance due to high mortality and increased costs of care and duration of hospitalizations. Azoles, primarily fluconazole, are the primary drugs utilized for these infections today, especially for invasive candidiasis and VVC, including recurrent VVC. However, medical guidelines are recommending that echinocandins be used for invasive candidiasis due to increasing resistance of azoles, especially fluconazole. In invasive candidiasis, VVC and recurrent VVC, infections due to non-albicans are increasing and are more likely in patients reporting current antibiotic use and coinciding with routine use of oral fluconazole. C. glabrata is intrinsically more resistant to fluconazole. Studies have reported as high as 50% of Candida species are non-albican for invasive candidiasis and as low as 10-20% for VVC. There is a need for alternative therapies, especially oral agents, given these growing statistics. SCY-078 is active against azole-resistant strains of Candida and by virtue of its fungicidal activity against Candida, it may provide a benefit in the treatment of invasive candidiasis, VVC and recurrent VVC. If approved, we believe SCY-078 would be positioned as the first and only oral and IV non-azole alternative for the treatment of fungal infections in hospital and outpatient settings. SCY-078 would particularly address the need of an oral agent for those patients infected with azole-resistant strains. Considering its activity against multidrug-resistant strains, SCY-078 is targeted for positioning as a safer and effective therapy for multidrug-resistant Candida strains with the flexibility of oral and IV administration. |
| |
• | Alternative and complementary to echinocandin regimens. Echinocandins are currently the recommended initial therapy for invasive candidiasis, but echinocandins can only be administered intravenously. Discharge from hospital with step-down to an oral azole risks relapse of an azole-resistant infection if the original pathogen was not identified and susceptibility determined. This leads some physicians to keep patients on IV echinocandins for the full course of therapy. If approved, SCY-078 would be positioned as the optimal step-down option after initial IV echinocandin therapy in patients with invasive candidiasis, allowing them to complete the full treatment with a glucan synthase inhibitor rather than switching to azoles. The IV-to-oral step-down within a single therapeutic class may allow earlier patient discharge from the hospital, resulting in reduced exposure to the risk of hospital-acquired infections and reduced costs of care. The IV formulation of SCY-078 would be positioned as an alternative treatment option to echinocandins, particularly relevant for those patients with infections due to echinocandin-resistant Candida. |
| |
• | Alternative to second generation azoles for Aspergillus. The IV and oral formulations of SCY-078 would be positioned as an alternative to currently available treatment options, such as voriconazole or isavuconazole, when treating azole-resistant Aspergillus infections or when the treatment with a glucan synthase inhibitor with fewer drug-drug interactions than azoles would be desirable. |
| |
• | Alternative to polyenes for treatment of multi-drug-resistant Candida. The only antifungal alternative currently available to treat invasive Candida spp. infections due to suspected or known azole- and echinocandin-resistant strains are the polyenes, such as Amphoteracin B, which are only available intravenously and have significant toxicities associated with their use. The IV and oral formulations of SCY-078 would be positioned as a safer alternative to Amphoteracin B when treating multi-drug-resistant strains. |
SCY-078 Development
We are developing both IV and oral formulations of SCY-078. Patients with invasive fungal infections are typically prescribed IV treatment in hospitals and then are switched, or “stepped down,” to oral formulations to complete their antifungal treatment after they have shown sufficient improvement. The duration of the entire antifungal regimen (IV plus oral) varies depending on the response to the antifungal treatment and the type of infection. Invasive candidiasis should be treated for at
least 2 weeks after negative cultures are obtained and invasive aspergillosis is typically treated for six to 12 weeks. The availability of SCY-078 in both oral and IV formulations would allow for maximum flexibility in the administration of the same agent during the entire antifungal regimen. The IV formulation would allow initiation of treatment in critically ill patients for whom IV therapy is preferred. The oral formulation would allow initiation of therapy in outpatient settings for those conditions that do not require hospitalization, such as VVC and recurrent VVC, as well as step-down from the initial IV antifungal agent (either SCY-078 or other glucan synthase inhibitor such as echinocandins), to complete the antifungal regimen, as indicated.
SCY-078’s clinical development program to date includes:
| |
• | Eight completed Phase 1 studies with the oral formulation. |
| |
◦ | SCY-078 was shown to be sufficiently safe and well-tolerated in eight completed Phase 1 studies supporting the progression of the development program. In a recently completed Phase 1 study the citrate salt of SCY-078 was shown to be well tolerated and resulted in a comparable pharmacokinetic (PK) profile to the phosphate salt, used in our previous clinical investigations. The citrate salt offers potential formulation development advantages and is being used in the IV formulation. Further development activities for both the oral and IV formulations of SCY-078 are planned with the citrate salt. |
| |
• | Two ongoing Phase 2 studies with the oral formulation. |
| |
◦ | We are currently conducting a multicenter Phase 2 study with primary endpoints of safety, tolerability, and pharmacokinetics of the oral formulation of SCY-078 as step-down treatment in patients initially treated with echinocandin therapy for invasive Candida infections, which are serious and life-threatening infections. We have opened new investigational sites in the U.S. and in Latin America and we are in the process of opening more sites in these regions and in Europe. Based on the data collected on the enrolled patients, together with the data from our recently completed Phase 1 biocomparison study, we expect to achieve the primary objectives of the study with fewer patients than originally planned and to report top line data by the end of the second quarter of 2016. |
| |
◦ | We are also conducting a multicenter Phase 2 study with primary endpoints of safety and efficacy of the oral formulation of SCY-078 in patients with VVC. We expect to complete the study and to report top line data by the end of the second quarter of 2016. We expect the data from this study to provide a confirmation of the potential therapeutic effect of orally administered SCY-078 in a clinical condition caused by Candida spp. and, along with the other clinical and nonclinical data from ongoing and planned activities, will contribute to the package of information that will support subsequent phases of development. |
| |
• | One ongoing Phase 1 study with an intravenous formulation of SCY-078. |
| |
◦ | We are conducting a single-rising-dose Phase 1 study to evaluate the safety, tolerability and pharmacokinetics of SCY-078 administered as an intravenous infusion in healthy subjects. We expect to complete the study and to report results by the end of the second quarter of 2016. |
Our Corporate Strategy
Key elements of our strategy include:
| |
• | to further develop SCY-078 and obtain regulatory approval in major commercial markets for our three key indications: invasive candidiasis, recurrent VVC and invasive aspergillosis; |
| |
• | to commercialize SCY-078 for selected indications in the U.S. through a dedicated sales force; |
| |
• | to contract with commercial partners to develop and commercialize SCY-078 outside of the U.S.; |
| |
• | to assess external opportunities to expand our clinical pipeline; and |
| |
• | to leverage our strong scientific team to pursue the development of other proprietary compounds. |
Overview of the Antifungal Market
Background of Invasive Fungal Diseases
Candida and Aspergillus species are responsible for approximately 85% of all invasive fungal infections in the United States and Europe. Infections caused by Candida rank as the fourth most common hospital-acquired bloodstream infection in the United States. There are approximately 400,000 cases of invasive Candida infections annually worldwide, including approximately 98,000 cases in the U.S. Invasive Candida infections result in a mortality rate ranging from 20% to 40%
depending on the immune status of the patient. Globally, an estimated 200,000 patients develop invasive Aspergillus infections annually, including approximately 46,000 in the U.S., and the mortality rate ranges from approximately 40 to 50%, even with treatment.
Hospital-acquired fungal infections due to Candida and Aspergillus species are becoming an increasing problem for the healthcare system. The increases in invasive fungal infections are due to the increased use of immune-suppressing chemotherapies and transplant drugs, and in-dwelling catheters, among other factors. Confirmed cases of invasive Candida infections rose in the United States by 52% between 2000 and 2005. In addition, the increase in use of broad spectrum antibiotics has been shown to contribute significantly to the risk of developing invasive fungal infections. Confirmed cases of invasive Aspergillus infections nearly doubled in the United States among patients receiving hematopoetic stem cell transplants between 2002 and 2005.
We believe confirmed cases of Candida blood infections account for only approximately one-quarter to one-third of Candida treatments. We further believe initiation of therapy prior to diagnosis, based on the presence of symptoms, represents a majority of the non-confirmed Candida treatments. This “empiric” therapy is clinically warranted because invasive Candida infections can be difficult to diagnose and the treatment outcome could be compromised by delaying initiation of therapy until the diagnosis is confirmed. Initiation of therapy within the first 12 hours following suspicion of fungal infection reduces the risk of death threefold. In addition, increased numbers of patients are undergoing procedures, such as chemotherapy,or solid organ or stem cell transplants, that cause or result in immune-suppression and therefore put patients at high risk of invasive Candida infections. As a result, the market opportunity for the treatment of invasive candidiasis is estimated to be greater than the number of confirmed cases reported for this condition.
The typical duration of antifungal treatment in cases of invasive candidiasis is around three weeks, and during the first 5-7 days an IV agent is recommended. If the condition of the patient improves, a step-down to oral treatment is considered after the initial IV therapy for the remainder of the antifungal regimen.
The recommended duration of antifungal treatment in cases of invasive aspergillosis varies with the response to antifungal therapy, ranging from six to 12 weeks in most cases. Typically, the antifungal agent is initiated intravenously and stepped down to an oral administration to complete the antifungal regimen. Because of the long duration of the antifungal regimen in aspergillosis and the fact that only azoles have oral formulations available, there are significant limitations in terms of flexibility of treatment options, particularly important for the treatment of azole-resistant cases.
Background of Vulvovaginal Candidiasis
Vulvovaginal Candidiasis, commonly known as a "yeast infection," is usually caused by Candida albicans and typical symptoms include pruritus, vaginal soreness, irritation and abnormal vaginal discharge. An estimated 75% of women of reproductive age will have at least one episode of VVC during their lifetime and 40-45% will experience two or more episodes. As many as 7-9% of these patients suffer from recurrent VVC, defined as experiencing at least four episodes during a 12-month period. Current treatments for VVC include topical antifungals and the use of prescription oral antifungals such as fluconazole, which has a therapeutic cure rate of 55% as reported in its label. We estimate there are approximately 370,000 recurrent VVC cases annually in the U.S. There are no products currently approved for the treatment of recurrent VVC.
Current Therapeutic Options
Fungal infections are currently treated using three main classes of antifungal drugs that target fungal cell membranes or cell walls. Each of these antifungal drugs has its own limitations that reduce its clinical usefulness.
Polyenes. Polyenes disrupt fungal cell membranes. The primary commercial polyene, amphotericin B, is used to treat a wide variety of fungi, including rare and difficult-to-treat species. However, polyenes have serious side effects including acute, potentially fatal kidney and heart injury, and they are only available intravenously. As a result, polyenes are typically used as a drug of last resort for treating invasive Candida and Aspergillus infections. Despite its toxicity, worldwide peak annual sales of lipid amphotericin B alone were approximately $379 million.
Azoles. Azoles, which block biosynthesis of a fungal cell membrane component, are the most frequently used class for treatment of invasive fungal infections and are the only class available in both IV and oral formulations. Azoles are used extensively for prevention and in unconfirmed cases. Fluconazole is recommended as the first-line therapy for acute uncomplicated VVC. However, while azole-sensitive species have been well-treated, this has permitted azole-resistant infections, with species such as Candida glabrata, to become more prevalent. Further, cross resistance among the azoles exists, which means that once an azole has been tried and failed, another azole may not be effective. Despite these limitations, fluconazole, the most commonly used azole, had worldwide peak annual sales of $1.1 billion and voriconazole, which is used for treatment of invasive aspergillosis, had worldwide peak annual sales of $685 million. The Infectious Disease Society of America (IDSA) guidelines for the treatment of invasive candidiasis have recently changed their recommendations and no longer recommend azoles as the initial treatment for most cases of invasive candidiasis. Azoles with activity against molds, such as voriconazole, are recommended as the first-line therapy for most cases of aspergillosis, but it should be noted that the
reports of azole-resistant Aspergillus strains have increased over the past years, mostly due to extensive use of azoles across medical settings, and in agricultural where they are used to protect crops against fungi.
Echinocandins. Echinocandins block biosynthesis of fungal cell walls by inhibiting a glucan synthase enzyme, an enzyme not found in human cells. The clinical success of echinocandins, particularly in azole-resistant infections, combined with their good tolerability profile, has resulted in these compounds being increasingly used in the treatment of invasive Candida infections. They are currently the recommended antifungal class for treatment of most cases of invasive candidiasis by the Infectious Disease Society of America (IDSA). However, echinocandins are only available in IV formulation. To allow for discharge from the hospital as quickly as possible, preferred medical practice is to transition eligible patients from IV to oral therapy. Without the availability of an oral echinocandin, physicians need to choose between administering oral azoles as a step down therapy, or keeping patients on an IV echinocandin therapy, which may require continued hospitalization. In addition, there are indications that resistance to echinocandins is emerging and being reported. Despite the limitations from being an IV-only therapy and having a special warning in Europe, Micafungin had worldwide peak annual sales of $326 million. Caspofungin, an IV-only therapy and the leading echinocandin, had worldwide peak sales of $530 million.
Antifungal Drug Resistance
Broad use of azole drugs has resulted in an increasing incidence of drug resistant Candida infections. At hospitals performing medically intensive procedures such as transplantation, rates of reduced azole susceptibility have reached 15-20%. We believe the rising level of azole resistance is driven by the reduction in prevalence of susceptible species such as Candida albicans and the resultant increase in prominence of infections caused by species inherently resistant to azoles, such as Candida glabrata and Candida krusei. Declining azole efficacy in Candida infections has led to echinocandins emerging as the drugs of first choice for treatment of most patients with invasive Candida infections. However, a recent study reported echinocandin resistance for Candida glabrata at an incidence rate exceeding 10%. Of the echinocandin resistant strains, the majority are also resistant to azoles, making these strains multi-drug resistant. Invasive Candida spp. infections due to suspected or known azole-resistant strains are typically treated with echinocandins for the entire duration of the treatment regimen. Because these agents are only available for IV administration, there is no opportunity for an effective step down strategy that would minimize hospital stays, hospital-acquired infections and associated costs to the healthcare system. The only antifungal alternative to treat invasive Candida spp. infections due to suspected or known azole- and echinocandin-resistant strains are the polyenes, which are only available intravenously and have significant toxicities associated with their use.
Broad use of azole drugs in the medical setting as well as in the agricultural setting has also fostered selection for azole resistance in Aspergillus species. Acquired resistance to azoles in Aspergillus fumigatus (the most common species associated with human infections) has become a public health concern. The frequency of azole-resistance in Aspergillus species has been reported ranging from 4% to 50% at different institutions.
A recent surveillance study that investigated the prevalence of azole-resistance in clinical Aspergillus isolates in an in-ternational network of centers indicated that azole-resistance is widely spread across most countries. Approximately 5% of the isolates from invasive aspergillosis cases showed resistance to azoles.
Our Product Candidate: SCY-078
SCY-078 Overview
We discovered and developed SCY-078 through a research collaboration with Merck Sharp & Dohme Corp., or Merck, a subsidiary of Merck & Co., Inc., and in May 2013 we acquired worldwide rights to SCY-078 in the field of human health. The compound is derived, by chemical modification, from enfumafungin, a natural product, and shows antifungal activity against Candida and Aspergillus through inhibition of glucan synthesis, similar to the echinocandin class. SCY-078 was shown to exhibit fungicidal activity against Candida albicans, the most common cause of invasive fungal infections among the Candida species. In addition, SCY-078 has shown potent in vitro activity against approximately 650 laboratory and clinically important strains of Candida and Aspergillus, including strains that are resistant to azoles and echinocandins. Activity against the majority of echinocandin-resistant strains suggests that SCY-078 represents a new class of antifungal agents (a novel glucan synthase inhibitor) that acts on a validated antifungal target in a manner distinct from the echinocandins.
In animal models of invasive fungal infections used to test other drugs that have proven to be effective in humans, SCY-078 was shown to be highly active against Candida spp. These studies allowed for the determination of the drug concentrations in blood required to achieve efficacy. These correlations of drug exposure to drug activity, or PK/PD, have been used to identify the predicted human exposure of SCY-078 believed to be required to achieve efficacy.
In Phase 1 studies, the oral formulation of SCY-078 has been shown to be sufficiently safe and well-tolerated in approximately 124 healthy human subjects at initial single oral doses of up to 1600mg in one day and doses of up to 800mg per day for 28 consecutive days to support progression into Phase 2 studies. Furthermore, oral dosing of the compound results in sustained blood concentrations in the range predicted from preclinical PK/PD studies to be required for efficacy.
We are currently conducting two Phase 2 studies with our oral SCY-078 formulation. The first study is evaluating the safety, tolerability, and pharmacokinetics of the orally administered SCY-078 as step-down treatment in patients initially treated with IV echinocandin therapy for invasive Candida infections. The second study is evaluating the safety and efficacy of orally administered SCY-078 for the treatment of vulvovaginal candidiasis.
We have also developed an IV formulation of SCY-078 that is currently being evaluated in a Phase 1 program.
In connection with our acquisition of the worldwide rights to SCY-078 in May 2013, Merck transferred to us responsibility for the investigational new drug application, or IND, for SCY-078, including all related technical documents, preclinical data, data from the seven Phase 1 trials conducted by Merck, and drug product and drug substance.
The Generating Antibiotics Incentives Now Act, or GAIN Act, was enacted in July 2012 to encourage the development of novel anti-infective drugs in the face of increasing drug resistance. Before the passage of the GAIN Act, the FDA traditionally required sponsors of novel antifungal drugs to use non-life threatening fungal infections, such as esophageal Candida infections, for a proof-of-concept study in preparation for Phase 3 studies in invasive diseases. This approach added time and cost to the process of developing novel drugs for invasive fungal infections. In order to encourage the development of treatments for serious or life-threatening infections, the GAIN Act required the FDA to review and ensure clear guidelines for clinical development of antibacterial and antifungal drugs. In September 2013 we met with the FDA to discuss our development program. In response, the FDA recommended we proceed with a smaller scale Phase 2 study directly in patients with invasive Candida infections, our intended patient population, without first conducting studies of esophageal Candida infections. These changes, we believe, may significantly reduce the time and expense associated with progressing SCY-078 through Phase 2 and Phase 3 studies.
The FDA has designated both the oral tablet formulation and IV formulation of SCY-078 as Qualified Infectious Disease Products, or QIDPs, under the GAIN Act and has also granted Fast Track designation to both formulations for their respective indications. The FDA’s Fast Track drug development program is a process designed to facilitate the development and expeditious review of drugs to treat serious conditions and fill an unmet medical need. This designation allows companies to interact with the FDA review team frequently to discuss critical development issues such as study design, required safety data necessary to support approval, and structure and content of an NDA. Additionally, should the FDA determine that a Fast Track product may be effective after their preliminary evaluation of clinical data submitted by a sponsor, the FDA may also consider reviewing portions of a marketing application before the sponsor submits the complete application, known as a “rolling” NDA. If SCY-078 is approved for its proposed use and awarded five years of exclusivity as a new chemical entity, it will be eligible for a ten-year period of data exclusivity, comprising five years of new chemical entity exclusivity plus an additional five years as a designated QIDP. This exclusivity period should protect SCY-078 from being subject to a competing abbreviated new drug application, or ANDA, for a generic drug, or from a competing 505(b)(2) new drug application for a follow-on product until the expiration of the exclusivity period.
SCY-078 is protected by an issued composition of matter patent in the United States, which expires in 2030. An additional composition of matter patent related to SCY-078 salt composition and polymorphs has been filed and is pending issuance. If granted, the new patent will extend the patent protection of SCY-078 salts, including the citrate salt currently under development, up to 2035. We have licensed rights to develop and commercialize SCY-078 in the field of human health in Russia and certain smaller non-core markets to R-Pharm, CJSC, or R-Pharm, a leading supplier of hospital drugs in Russia, in exchange for an upfront payment, royalties, and their expertise and financial assistance in developing the compound, as more completely described under the heading “Collaborations and Licensing Agreements” as set forth below.
In summary, SCY-078 has unique attributes that define its potential to address significant unmet medical needs and market opportunities including:
| |
• | only glucan synthase inhibitor (GSI) with both oral and IV formulations in clinical development, allowing first-line treatment and oral step down in the same class. |
| |
• | distinct chemical structure from other GSIs, allowing unique spectrum of activity and pharmacokinetic profile. |
| |
• | fungicidal capabilities against Candida species compared to azoles, which are fungistatic |
| |
• | activity against azole- and most echinocandin-resistant Candida strains; potentially providing a safer alternative to amphotericin B for multi-drug-resistant Candida infections. |
| |
• | activity against azole-resistant Aspergillus strains. |
SCY-078 Target Product Profile
We believe that there are significant commercial opportunities for a new antifungal drug that has potent activity against azole and echinocandin susceptible and resistant Candida and Aspergillus strains, is available in both oral and IV formulations,
and has a favorable safety and tolerability profile. If approved, SCY-078 has the potential to address significant gaps with commercially available therapies and could be used as follows:
Treatment of invasive Candida infections. If SCY-078 is approved for the treatment of invasive Candida infections, we believe it could replace echinocandins as the drug of choice for these infections because it would be available in both IV and oral forms. An orally effective non-azole antifungal with the preferred mechanism of glucan synthase inhibition would allow patients to be transitioned more easily from a hospital-based care to outpatient care, which may reduce, or eliminate, expensive hospital stays and risks of hospital-acquired infections. Additionally, SCY-078 could be used as the step-down therapy from any echinocandin, replacing fluconazole and providing the advantage of continuing the antifungal treatment with an oral glucan synthase inhibitor.
Treatment of infections due to drug-resistant Candida. SCY-078 has been shown to be effective preclinically against Candida species resistant to azoles, including Candida albicans, Candida glabrata and Candida krusei. In addition, SCY-078 has been shown to be effective in vitro against the majority of echinocandin-resistant Candida strains tested. SCY-078 has the potential to provide a first-line treatment against invasive Candida infections, including those known or suspected to be resistant to currently available azoles and echinocandins.
Treatment of invasive Aspergillus infections. If SCY-078 is approved for the treatment of invasive Aspergillus infections, we believe it could offer significant advantages over the current first-line azole therapy of voriconazole due to the numerous drug interactions and adverse events associated with the use of voriconazole. Furthermore, SCY-078 has been shown to be effective in vitro against all azole-resistant strains of Aspergillus tested. SCY-078 could provide a first-line treatment against invasive Aspergillus infections known to be resistant to currently available azoles.
Prevention of Candida and Aspergillus infections. If SCY-078 is approved for use as a preventative treatment for Candida and Aspergillus infections, SCY-078 has the potential to offer advantages over current prophylactic drugs because of its activity against fungal strains that are resistant to azoles.
Treatment of vulvovaginal candidiasis (VVC): If SCY-078 is approved, it could provide a first-line therapy for recurrent VVC, for which there is no treatment currently approved. Fluconazole administered for up to six months is recommended for recurrent VVC, but recurrences are observed rapidly after stopping fluconazole due to the persistence of the fungi in the vaginal reservoir. In contrast with fluconazole that is fungistatic against Candida spp., SCY-078 is fungicidal against most Candida isolates. We believe this “cidal” activity (i.e., killing the pathogen) would provide an advantage in preventing recurrences. SCY-078, with its activity against azole-resistance strains, may also be adequate to treat VVC episodes caused by azole-resistant isolates.
Preclinical Characterization of SCY-078
SCY-078 has broad antifungal activity based on a proven mechanism of action
SCY-078 is a potent inhibitor of the synthesis of the polymer beta 1,3 D glucan, an essential component of the fungal cell wall of Candida and Aspergillus species. Glucan synthesis inhibition is a clinically proven antifungal mechanism, as demonstrated by the echinocandin class of antifungal agents. Activity of SCY-078 observed against the majority of echinocandin-resistant strains suggests that SCY-078 acts in a manner distinct from the echinocandins.
SCY-078 is active in vitro against a broad spectrum of Candida and Aspergillus species
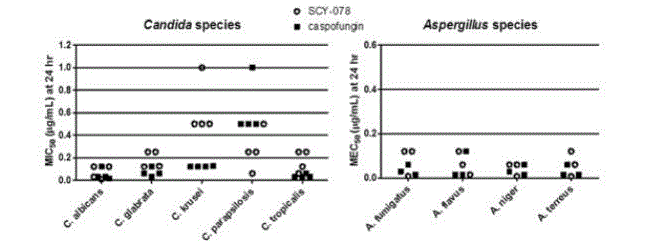
SCY-078 has been shown to have potent activity in vitro against over 500 strains from eleven Candida species and 150 strains from four Aspergillus species. The charts below summarize the in vitro activity of SCY-078 against a collection of “wild-type” strains (i.e., those having no known drug resistance) of Candida and Aspergillus. Drug activity was measured as the minimum concentration of drug that inhibits replication of Candida or growth of Aspergillus by more than 50% relative to untreated cultures (MIC50 and MEC50, respectively). Each data point represents the average activity value for all strains tested at a single laboratory. Four laboratories were used for evaluation of Candida and three laboratories were used for evaluation of Aspergillus to confirm reproducibility of results among independent test sites. The potency of SCY-078 against these Candida and Aspergillus strains is comparable, within assay variability, to that of Caspofungin, the current leading echinocandin.
SCY-078 is active in vitro against azole-resistant Candida and Aspergillus strains
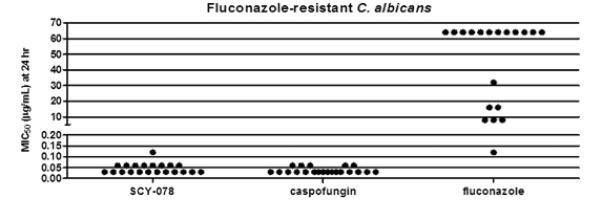
Widespread use of azole drugs has resulted in azole-resistant strains of Candida and Aspergillus becoming increasingly prevalent, leading to treatment failures. Cross resistance among azoles means that once an azole has been tried and failed, another azole may not be effective. SCY-078 was active against all azole-resistant Candida strains tested, with activity comparable to that observed against wild-type strains. As shown in the graph below, the in vitro activity of SCY-078 was comparable to that of Caspofungin, the leading echinocandin, against Candida albicans resistant to fluconazole, a leading azole.
SCY-078 was also active against all azole-resistant Aspergillus strains tested, with the range of MEC50 values comparable to those observed against wild-type strains.
SCY-078 is active in vitro against a majority of echinocandin-resistant Candida species
Echinocandin resistance is increasing in prevalence, particularly among azole-resistant species such as Candida glabrata. As illustrated in the figure below, SCY-078 retained in vitro activity against a majority of echinocandin-resistant Candida glabrata strains tested when defined as minimum inhibitory concentrations (MICs) similar to those determined for wild type Candida. Similar results were observed for echinocandin-resistant strains of other Candida species. Thus, SCY-078 may offer a therapeutic option against multi-drug resistant strains such as those that have emerged in Candida glabrata.
Nonclinical toxicology studies determined safety parameters to monitor in SCY-078 in clinical studies
The preclinical safety of SCY-078 has been evaluated in multiple exploratory and GLP, or Good Laboratory Practice, studies in rats, dogs, rabbits, and non-human primates. The longest duration of oral dosing has been 28 days and the longest duration of IV dosing has been 14 days.
In these studies, at the highest tested doses, at exposures seven-fold the targeted efficacious exposure, very slight to moderate toxicities were observed in two animal species. The two major organs impacted were the stomach (degeneration of the stomach lining) and the liver (single cell necrosis). In rats, the degeneration of the stomach lining was reversible after cessation of dosing. Degeneration of the stomach lining observed in preclinical toxicology studies was not seen in healthy subjects in the Phase 1 multiple dose study where individuals who received 800mg SCY-078 daily for 28 days had pre- and post-treatment endoscopy with gastric biopsy. The results from the completed 14-day toxicity studies in rats and dogs conducted with an IV formulation of SCY-078 demonstrate that the toxicity profile of an IV formulation of SCY-078 is similar to the toxicity profile previously observed in the non-clinical safety program for the oral formulation. Infusion site reactions were observed after IV administration of SCY-078. These reactions more prominent at the highest dose tested in rats and rarely reported in dogs. Considering the overall tolerability of the IV formulation in both species, the toxicology program was considered adequate by the FDA to support the initiation of the first Phase 1 study with an IV formulation of SCY-078. In preliminary developmental and reproductive toxicity studies, SCY-078 did not cause any developmental toxicity in two animal species up to the maximum tolerated dose. In safety pharmacology studies, there were no clinically significant effects of SCY-078 on markers of cardiovascular, respiratory or central nervous system function.
Toxicology studies are planned to enable oral dosing for up to 90 days and IV administration for up to 28 days. Additional reproductive toxicity studies are also planned to support subsequent stages of development.
Should additional optimization be needed for the IV formulation, additional toxicology studies will be conducted to evaluate the safety and tolerability of any potential changes in the IV formulation.
Preclinical pharmacokinetic and drug metabolism properties of SCY-078 support effective oral administration and limited drug-drug interactions
SCY-078 has been evaluated broadly in preclinical pharmacokinetic and drug metabolism studies at exposure levels that were higher than those expected to be required to effectively treat infections in humans. SCY-078 was orally bioavailable in all four animal species studied.
Many patients with, or at risk of, invasive fungal infections are taking other medications, making it important to consider drug-drug interactions. The leading azoles have significant effects on the metabolism of many medications, which can lead to over-dosing or toxicity from co-administration of drugs. In vitro, SCY-078 interacts with few drug metabolizing enzymes, and does not induce CYP3A4 (the major drug metabolizing enzyme), thus it may cause fewer clinically relevant drug-drug interactions. The propensity for SCY-078 to be involved in drug-drug interactions will be studied in Phase 1 clinical studies.
In vivo animal studies predict that SCY-078 can be effective in treating invasive fungal infections
Mouse models of Candida and Aspergillus infections have been predictive of clinical efficacy for the approved glucan synthesis inhibitors. SCY-078 was evaluated in multiple studies in Candida albicans-infected mice. In these studies, SCY-078 treated animals had no measurable Candida in organs tested following doses which resulted in drug levels in the blood similar
to those that have been safely achieved in humans. Comparable results were observed in mice infected with other Candida species, including Candida glabrata.
The in vivo efficacy of SCY-078 was also evaluated against Aspergillus fumigatus in multiple studies. When infected with Aspergillus, mice with partially deficient immune defenses develop aggressive infections that generally result in death. However, SCY-078-treated mice exhibited dose-dependent increases in survival rates up to 90%, as measured in the first 21 days after infection.
In summary, SCY-078 demonstrated potent in vivo antifungal activity in mouse models of Candida and Aspergillus infection studied, supporting our expectation of clinical efficacy for SCY-078.
Clinical Experience with SCY-078
To date, eight Phase 1 safety and pharmacokinetic studies have been completed using the oral formulation of SCY-078. Four of the eight studies evaluated a single oral dose while four evaluated multiple oral doses of SCY-078.
SCY-078 consistently showed sufficient safety and tolerability in Phase 1 studies to support progression into Phase 2 studies.
Approximately 124 healthy subjects have received at least one dose of oral SCY-078 in Phase 1 studies. SCY-078 was generally well tolerated at initial single oral doses of up to 1600mg in one day and doses of up to 800mg per day for 28 consecutive days. The majority of reported adverse events have been generally transient and primarily mild to moderate in intensity.
The preliminary safety and PK data from the completed Phase 1 studies are summarized in the following table:
|
| | | | | | | | |
Design/Objective | | Clinical Endpoints | | Subject Population | | Dosing Regimen | | Results |
| | | | |
Phase 1, randomized, double-blind, placebo-controlled, single ascending-dose, safety, tolerability, and PK study | | Safety and tolerability by physical examination, vital signs, ECGs and laboratory safety evaluations (hematology, chemistry, urinalysis), gastrin levels; PK data in fasted state and after high fat meal | | 16 healthy males (18–45 years) | | Panel A: 8 subjects: single doses 10, 40, 150, 600, and 1600mg SCY-078 (6 active / 2 placebo for each dose) Panel B: 8 subjects: single doses 20, 80, 300, and 800mg SCY-078 (6 active / 2 placebo for each dose) | | Safety: SCY-078 up to 1600mg was generally safe and well tolerated; no serious adverse events (SAEs) reported. Statistical analysis of PK parameters [AUC (“area under the curve”, a measure of cumulative drug exposure over a defined post-dose time interval), Tmax (time of maximum circulating drug concentration) and Cmax (maximum circulating drug exposure)] indicated that: 1) Dose proportionality was observed for doses up to 1600 mg 2) Dosing SCY-078 drug-filled capsules with a high fat meal increased drug exposure levels by ~20% compared to levels observed in fasted subjects, which was within intersubject variability |
| | | | |
Phase 1, double-blind randomized, single dose study to evaluate the safety, tolerability, and PK in elderly subjects | | Safety and tolerability by physical examination, vital signs, ECGs and laboratory safety evaluations (hematology, chemistry, urinalysis); PK data | | 17 healthy males and females (65–85 years) | | Panel A: 500 mg SCY-078/Placebo Panel B: Placebo/500 mg SCY-078 (6 active / 2 placebo for each panel) | | Safety: SCY-078 generally well tolerated. One non-drug -related SAE of metastatic carcinoid tumor was reported. The most common adverse events (AEs) were gastrointestinal disorders and nervous system disorders. Statistical analysis of PK parameters (AUC, Tmax and Cmax) indicated that exposure levels were ~30% higher in elderly patients compared to young males. |
| | | | | | | | |
Phase 1, Open label biocomparison study of two formulations of SCY-078 and a pantoprazole interaction study with SCY-078 in healthy subjects | | Safety, tolerability and PK of fit-for-purpose (FFP) drug filled capsules compared to FFP compressed tablets; impact of multiple doses of a proton pump inhibitor on single doses of SCY-078; impact of high fat meal on FFP compressed tablets | | 16 healthy males (18–45 years) | | Periods 1 and 2: Single doses of 500 mg SCY-078 (as five 100mg FFP dry filled capsules or two 250mg FFP compressed tablets) Period 3: Pantoprazole 40mg X 5 days and 500 mg SCY-078 (two 250mg FFP compressed tablets) Period 4: 500 mg SCY-078 (two 250mg FFP compressed tablets) administered after a high fat meal | | Safety: SCY-078 generally well tolerated. One SAE of elevated liver enzymes that led to discontinuation was reported. The most common AEs were gastrointestinal disorders. Statistical analysis of PK parameters (AUC, Tmax and Cmax) indicated that: 1) Exposure levels in patients who received compressed tablets were ~20% higher than in those who received drug filled capsules 2) Exposure levels of SCY-078 in patients were approximately 25% lower when administered with the proton pump inhibitor pantoprazole compared to SCY-078 administered alone 3) Dosing SCY-078 tablets with a high fat meal increased drug exposure levels by ~50%–60% compared to levels observed in fasted subjects |
| | | | | | | | |
|
| | | | | | | | |
Design/Objective | | Clinical Endpoints | | Subject Population | | Dosing Regimen | | Results |
| | | | |
Phase 1, randomized, double-blind, placebo-controlled, multiple ascending-dose safety, tolerability and PK study | | Safety and tolerability by physical examination, vital signs, ECGs and laboratory safety evaluations (hematology, chemistry, urinalysis), gastrin levels and gastric histology; Plasma PK data and concentrations of intact drug in urine after multiple doses of SCY-078 | | 32 healthy males (18–45 years) | | 300, 600, and 800 mg SCY-078 or matching placebo once daily for 10 days, or 800 mg SCY-078 or matching placebo once daily for 28 days. (6 active /2 placebo in each panel) | | Safety: SCY-078 was generally safe and well tolerated. Most common AEs were headache, lack of energy, dizziness, and gastrointestinal disorders. Statistical analysis of PK parameters (AUC, Tmax and Cmax) indicated that: 1) The target drug exposure level (AUC of 17µM.hr) was approached after 10 days of dosing at 600mg per day 2) Two weeks were needed to reach steady state concentrations in many subjects 3) Exposure levels were ~2.3 fold (Cmax) to 3.3 fold (AUC) higher after 26 days of dosing compared to the first day Insignificant concentrations of SCY-078 were found in urine. |
|
| | | | | | | | |
| | | | |
Phase 1, randomized, partially-blind, placebo-controlled study of multiple doses of ketoconazole on single dose PK of SCY-078 | | Safety and tolerability of SCY-078 Single dose PK profile of SCY-078 after multiple doses of ketoconazole | | 14 healthy males (18–45 years) | | Period 1: 100 mg SCY-078 or matching placebo Period 2: Ketoconazole 400 mg once daily for 15 days starting on Day -1 with a single dose of 100 mg SCY-078 (or placebo) coadministered on Day 1. 12 Subjects (10 active / 2 placebo) | | Safety: SCY-078 was generally well tolerated when dosed alone or with ketoconazole. The most common AEs were headache and increased ALT/AST. Statistical analysis of PK parameters (AUC, Tmax and Cmax) indicated that in the presence of ketoconzaole 1) Drug exposure as measured by AUC was ~5.7 fold higher 2) Cmax increased 2.5 fold |
| | | | |
Phase 1, randomized, double-blind, placebo controlled multiple dose study to assess the safety, tolerability, and PK of a loading dose of SCY-078 | | Safety and tolerability of SCY-078; PK profile of SCY-078 after a loading dose on day 1 | | 8 healthy males (18–45 years) | | 1800 mg SCY-078 (or placebo) administered as 600 mg TID (three times a day) on Day 1, followed by 500 mg SCY-078 (or placebo) QD (once daily) on Days 2-7. 8 Subjects (6 active / 2 placebo) | | Safety: SCY-078 was generally well tolerated. No SAEs or discontinuations. The most common AE was diarrhea; 1 subject had elevated bilirubin. Statistical analysis of PK parameters (AUC, Tmax and Cmax) indicated that the loading dose on day 1 achieved a target drug exposure (AUC of ~20.8µM.hr). Drug exposures observed under the QD maintenance dosing regimen were ~20.8µM.hr on Day 3 and ~16µM.hr on Day 7. |
| | | | |
Phase 1, open-label, fixed-sequence, multiple-dose study investigating the effect of diltiazem on the PK and safety of SCY-078 in healthy subjects | | Safety and tolerability of SCY-078; PK profile of SCY-078 after multiple doses of diltiazem | | 16 healthy males (20-45 years) | | Treatment A (Period 1), 200 mg SCY-078 q6h (total dose of 600 mg) on Day 1 and 100 mg SCY-078 QD Days 2 to 14. Treatment B (Period 2), 240 mg of diltiazem QD on Days 1 to 14, 200 mg of SCY-078 q6h (total dose of 600 mg) on Day 1, and 100 mg SCY-078 QD Days 2 to 14. | | Safety: SCY-078 generally well tolerated. The most common AE was headache. No SAEs; 1 discontinuation due to first degree heart block following administration of diltiazem only Statistical analysis of PK parameters (AUC, Tmax and Cmax) indicates that in the presence of diltiazem: 1) Drug exposures as measured by AUC were ~2.5 fold higher 2) Cmax was increased 2 fold |
| | | | | | | | |
A Phase 1, 3-Period, Open-Label, Oral Biocomparison Study of 2 Formulations of SCY-078 with a Food Effect Period in Healthy Subjects
| | Bioavailability comparison of 2 oral formulations and investigation of food effect.
| | 24 healthy males (18-50 years)
| | Treatment A = Oral doses of 500-mg SCY-078 phosphate salt formulation in the fasted state. Treatment B = Oral doses of 500-mg SCY 078 citrate salt formulation in the fasted state. Treatment C = Oral doses of 500-mg SCY 078 citrate salt formulation in the fed state (following a high-fat breakfast) | | Safety: SCY-078 was generally well tolerated. No SAEs were reported. One subject was discontinued due to increased ALT/AST; the event was considered non serious by the investigator and resolved without intervention. The most common AEs were mild or moderate nausea, abdominal pain and diarrhea. Analysis indicated that the overall PK profiles for the phosphate and citrate tablet formulations were very similar. Food increased the average Cmax for the citrate formulation by approximately 37% and average AUC by approximately 51%.
|
The most frequently reported adverse events have been gastrointestinal. In multiple dose studies, these included diarrhea, abdominal pain or discomfort, and vomiting. These gastrointestinal side effects were not considered serious in nature and only one subject discontinued dosing with SCY-078 when he withdrew consent due to gastrointestinal adverse events. In one study six subjects who received 800mg SCY-078 daily for 28 days underwent pre-treatment and end-of-treatment gastric endoscopy
with biopsy, with no evidence of stomach lining degeneration or other significant clinical finding observed. None of the 66 subjects receiving SCY-078 in the four Phase 1 studies in which serum gastrin levels were monitored exhibited levels outside the normal range.
Two subjects have experienced significant liver function test increases in the Phase 1 studies. The first subject was a 27-year old man who had elevated levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) (which are indicators of liver function and integrity that can be detected in typical laboratory tests), after a 500mg single dose of SCY-078. The event was deemed by the investigator as serious and to be study drug related. Other markers of liver injury remained within the normal range. ALT/AST levels decreased over the 48-hour period post-dose and this subject’s liver function tests returned to the normal range without intervention. A second subject, in a different study, was a 22-year old female who had an elevation of ALT and AST after a second dose of 500mg of SCY-078. The event was deemed by the investigator not be serious but to be study-drug related. The subject admitted alcohol drinking and acetaminophen intake at the time of enzyme elevation. Other markers of liver injury remained within the normal range. The elevated enzymes returned to normal without intervention.
No other serious adverse events deemed related to study drug have been reported in any of the Phase 1 studies completed to date.
SCY-078 exhibits favorable pharmacokinetic properties in humans
As a result of Phase 1 studies of SCY-078, we believe that SCY-078 can be sufficiently well absorbed as an oral medication to achieve the drug levels necessary to be effective in treating patients. The half-life of approximately 20 hours supports once daily dosing and a loading dose on day 1 should result in therapeutic concentrations being achieved on the first day of treatment. Drug exposure increased proportionally and in a predictable manner with doses up to the maximum dose tested (1600mg in single dose studies). There were no major differences in the pharmacokinetics or safety of SCY-078 in healthy elderly subjects relative to younger adults, an important consideration since many patients experiencing invasive fungal infections are elderly.
Results from clinical studies conducted thus far to determine the potential for clinical drug-drug interactions confirmed that SCY-078 can likely be used, with suitable dose adjustments, in combination with moderate inhibitors of the most common drug metabolizing enzyme (CYP3A). The drug interaction studies were performed with ketoconazole (strong inhibitor of CYP3A4) and diltiazem (moderate inhibitor of CYP3A4). Results of these studies indicate that a dose reduction of SCY-078 will be required with moderate CYP3A inhibitors and co-administration with strong inhibitors will not be recommended.
A drug interaction study was also conducted with pantoprazole, a proton pump inhibitor. In this study, SCY-078 concentrations with pantoprazole were ~25% lower than SCY-078 alone; the results met the hypothesis that exposures of SCY-078 with or without a proton pump inhibitor were similar. Other studies to evaluate the potential of drug-drug interactions with SCY-078 are planned.
A biocomparison study was conducted between drug filled capsules that were used in early Phase 1 studies and compressed tablets which will be used in future studies. Compressed tablets had concentrations that were ~20% higher than capsules. The effect of a high fat meal on SCY-078 when dosed as compressed tablets indicated exposures that were ~50 to 60% higher than when administered in a fasted state.
Our clinical data, together with mouse efficacy data, support therapeutic activity for SCY-078
Correlations of circulating drug levels to drug efficacy in preclinical mouse infection models can be translated into human patients and are an established tool in the development of antifungal drugs. The efficacious drug levels determined for SCY-078 in the mouse models indicate that the levels achieved in the human Phase 1 clinical trials are predictive of efficacy in infected patients. Specifically, in human subjects who received SCY-078 as a loading oral dose of 600mg three times per day (1800mg/day) followed by a maintenance daily dose of 500mg, the circulating levels of SCY-078 exceeded those that cured the infection in the mouse models of invasive Candida infections. These results indicate that SCY-078 can be administered to patients with invasive Candida infections at doses that are predicted to be effective and are generally well tolerated.
Current SCY-078 Clinical Development Activities
Based on results from studies to date, we believe that SCY-078 has the potential to offer a new therapeutic option to treat several fungal infections, including serious and invasive fungal infections. The goal of the clinical development plan for SCY-078 is to provide sufficient safety and efficacy data for submission and FDA approval of an NDA.
We anticipate that our initial NDA submission would seek approval for an indication for oral and IV formulations of SCY-078 for the treatment of invasive Candida infections (or invasive candidiasis). We expect additional Phase 3 and post-market studies, and supplemental NDAs, to expand the list of indications to include treatment of invasive Aspergillus infections, recurrent vulvovaginal candidiasis and prevention of invasive fungal infections.
Phase 1 studies of the Intravenous Formulation of SCY-078
We have initiated our first Phase 1 study with the IV formulation of SCY-078. The study is a double-blind, randomized, placebo-controlled, alternating panel, rising single intravenous dose study in healthy male subjects. It is designed to evaluate the safety, tolerability, and pharmacokinetics of single ascending doses of intravenously administered SCY-078 in healthy volunteers. The results from this study are expected by the end of the second quarter of 2016 and will be used to guide the design of subsequent studies with the IV formulation.
SCY-078 Phase 2 studies with the oral formulation of SCY-078
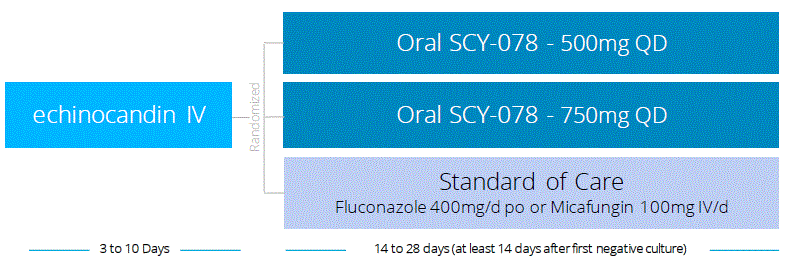
SCY-078 as an Oral Step-Down in the Treatment of Invasive Candida Infections: A Phase 2 study designed to evaluate the pharmacokinetics, safety and efficacy of oral SCY-078 as step-down therapy after IV echinocandin in patients with invasive Candida infections, is ongoing. This is a three-arm study comparing step-down oral therapy with two doses of SCY-078 to current standard of care based on current Infectious Disease Society of America Practice Guidelines. All patients will receive initial therapy with an IV echinocandin for three to 10 days. Based on clinical and microbiological response, patients will be switched to randomized therapy as illustrated in the figure below. Patients in arm one will switch to oral SCY-078 dosed at 1000mg on day one followed by once daily dosing of SCY-078 500mg. Patients in arm two will switch to oral SCY-078 dosed at 1250mg on day one followed by once daily dosing of SCY-078 750mg. Patients in arm three will receive standard of care. Current standard of care calls for a switch to oral therapy with fluconazole 400mg/day after loading dose of 800mg on day 1, unless the patient is infected with a Candida strain that is not susceptible to fluconazole, in which case the patient will be maintained on IV micafungin for the remainder of therapy. Antifungal treatment will be continued for at least 14 days after the first negative culture for Candida and resolution of signs and symptoms of infection.
We have opened new investigational sites in the U.S. and in Latin America and we are in the process of opening more sites in these regions and in Europe. Based on the data collected on the enrolled patients, together with the data from our recently completed Phase 1 biocomparison study, we expect to achieve the primary objectives of the study with fewer patients than originally planned and to report top line data by the end of the second quarter of 2016. The study’s identification number on www.ClinicalTrials.gov is NCT02244606.
SCY-078 as an oral therapy for Vulvovaginal Candidiasis (VVC): We are also investigating the potential clinical utility of SCY-078 in other areas of unmet medical need such as vulvovaginal candidiasis, or VVC, caused by Candida spp. VVC is a highly prevalent condition with limited therapeutic options for infections caused by azole-resistant Candida spp. We initiated a Phase 2 study evaluating the safety and efficacy of orally administered SCY-078 in this indication in the fourth quarter of 2015. This is a randomized, evaluator-blinded study that will enroll approximately 90 patients with an acute episode of moderate to severe VVC and a history of previous recurrences. The study will evaluate two dose regimens of oral SCY-078 and includes oral fluconazole as the active comparator arm. Under this study patients are randomized to receive either 1250mg of SCY-078 on day one followed by 750mg QD for two or four days or 150mg of fluconazole on day 1 (i.e., currently approved dose of fluconazole for this indication). The enrollment is progressing and we expect top line results by the end of the second quarter of 2016. We also expect the data from this study to provide a confirmation of the potential therapeutic effect of orally administered SCY-078 in a clinical condition caused by Candida spp. and, along with the other clinical and nonclinical data from ongoing and planned activities, to contribute to the package of information that will support subsequent phases of development. The study's identification number on www.ClinicalTrials.gov is NCT02679456.
Planned SCY-078 Clinical Development Activities
The following clinical studies are planned:
Phase 1 drug-drug interaction studies of SCY-078: Two additional drug-drug interaction studies are planned during 2016. The studies are intended to better define the potential effect of co-administration of SCY-078 with other drugs. The
design of these studies is currently under evaluation and will include drugs that are either often administered in the intended patient population or a representative of a particular metabolic pathway.
Phase 1 studies of the Intravenous Formulation of SCY-078: We are planning to conduct a multiple dose rising study to evaluate the safety, tolerability, and pharmacokinetics of an IV formulation of SCY-078 when administered to healthy volunteers for up to 14 days. If additional formulation optimization is deemed necessary, further single and/or multiple dose Phase 1 studies with an IV formulation may be needed.
SCY-078 (IV and Oral) for the Treatment of Invasive Candida Infections: The next study to evaluate the efficacy and safety of SCY-078 in patients with invasive Candida infections will include both the IV and oral formulations of SCY-078. We expect it to be a multicenter, randomized, double-blinded, active comparator, non-inferiority study evaluating the safety and efficacy of SCY-078 compared to standard of care. The current standard of care, per the Infectious Disease Society of America, is the initiation of antifungal therapy with an echinocandin (IV) and step-down to oral fluconazole for completion of the antifungal regimen. For infections caused by an azole-resistant Candida strain the standard of care is to administer an echinocandin (IV) for the entire duration of the antifungal treatment. The final design from this study will be informed by the results from our ongoing clinical investigations and subsequent discussions with relevant regulatory agencies such as the FDA. We are planning to initiate this study in the first half of 2017.
SCY-078 (IV and Oral) for the Treatment of Invasive Fungal Infections that are Refractory to or Intolerant of standard Antifungal agents : This study will evaluate SCY-078 in infections where there is unmet need and the potential to show differentiation from available therapies for invasive fungal infections for which SCY-078 may show potential clinical utility, including invasive candidiasis. This will be an open-label study in which SCY-078 will be administered to patients bearing invasive fungal infections that are refractory to, or the patients are intolerant of, standard therapy (azoles, echinocandins or polyenes). The final design of this study will be informed by our ongoing clinical and non-clinical investigations and subsequent discussions with relevant regulatory agencies such as the FDA. We are planning to initiate this study in the fourth quarter of 2016. It is possible that compelling data in this study could result in streamlined development to an initial NDA for a restricted indication.
SCY-078 (Oral) for the Treatment of Vulvovaginal Candidiasis (VVC): Subsequent stages of the development program for the VVC indication will be informed by the results from our ongoing Phase 2 study and will likely include a Phase 2b dose ranging study and two Phase 3 studies in the selected population.
Planned SCY-078 Non-Clinical Development Activities
We are planning additional non-clinical development activities to support our SCY-078 development program, including further characterization of the antifungal spectrum of activity and toxicology assessments to enable long term treatment durations.
Clinical Development Activities in Invasive Aspergillosis
We are encouraged by our existing in vitro and in vivo data indicating activity against a broad range of Aspergillus species, including drug-resistant strains. In the future, we expect to prepare a clinical development plan for SCY-078 as a therapy for invasive aspergillosis.
Commercialization, Marketing and Sales of SCY-078
Given our stage of development, we have not yet established a commercial organization or distribution capabilities.
We expect that prescribing physicians for the treatment of invasive fungal infections will be located at major medical centers, where physicians specializing in critical care, infectious disease specialists, and physicians treating immune-compromised or immune-suppressed patients, such as oncologists and those performing solid organ transplants and stem cell transplants, are likely to be found.
We intend to form our own focused hospital-based sales and marketing force to target physicians in the United States. Outside of the United States, subject to obtaining necessary marketing approvals, we likely will seek to commercialize SCY-078 through distribution or other collaboration arrangements. We have already entered into an agreement pursuant to which we licensed to R-Pharm rights to develop and commercialize SCY-078 in the field of human health in Russia and certain smaller non-core markets.
For VVC, we anticipate that prescribing physicians will mostly be obstetricians and gynecologists and likely a number of primary care physicians and, we believe, it will require a specific sales and marketing force with a women's health focus. We will assess our commercial strategy for VVC in the future.
Competition for SCY-078
Our competitors include large pharmaceutical and biotechnology companies, and specialty pharmaceutical and generic drug companies. The three leading branded antifungal drugs representing one from each main class are as follows:
Azoles. V-fend® (voriconazole), a product marketed by Pfizer (peak worldwide sales of $685 million). Merck also markets the azole Noxafil® (posaconazole) and Astellas has the marketing rights to isavuconazole, recently approved in the U.S. and other global markets;
Echinocandins. Cancidas® (Caspofungin), a product marketed by Merck (peak worldwide sales of $530 million). Pfizer also markets the echinocandin Eraxis® (anidulafungin) and Astellas markets the echinocandin Mycamine® (micafungin); and
Polyenes. AmBisome® (liposomal amphotericerin B), a product sold by Gilead in Europe, by Astellas in the U.S. and by Dainippon-Sumitomo in Japan (peak worldwide sales of $379 million).
Pfizer, Merck and Astellas are all large pharmaceutical companies with significant experience and financial resources in the marketing and sale of specialty pharmaceuticals. Various other producers market and sell generic oral voriconazole, fluconazole and itraconazole.
Further, we expect that product candidates currently in clinical development, or that could enter clinical development in the near future, may represent significant competition, if approved. These include the triazole VT-1161 being developed by Viamet Pharmaceuticals, Inc., the echinocandin CD101 being developed by Cidara Therapeutics, Inc., and the polyene MAT2203 developed by Matinas BioPharma Holdings Inc. In addition, several compounds that have novel methods of action are currently in various stages of development, including T-2307 developed by Toyana, F901318 developed by F2G Limited and VL-2397 being developed by Vical Inc. These companies may have significantly greater resources than ours.
The key competitive factors affecting the success of SCY-078, if approved, are likely to be its efficacy, safety, convenience, price, use in outpatient settings, the level of generic competition and the availability of reimbursement from government and other third-party payors. If approved, we believe that SCY-078’s features, including its oral dosing and efficacy against resistant strains, will differentiate it from competing products. We believe that SCY-078 will compete favorably against competing products in efficacy, safety, convenience and use in out-patient settings, allowing us to price SCY-078 at a premium to generics and other competing products.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than products that we may develop. Our competitors also may obtain FDA, or other regulatory, approval for their products more rapidly than we may obtain approval for ours. In addition, our ability to compete may be affected because in many cases insurers or other third-party payors seek to encourage the use of generic products. In the azole class, fluconazole, itraconazole, and oral voriconazole are generic. There is currently no generic echinocandin, but Caspofungin, the largest selling echinocandin, is expected to become available on a generic basis over the coming years and perhaps prior to the launch of SCY-078. If approved, we believe SCY-078 will be capable of delivering value supportive of premium pricing over competitive generic products.
Manufacturing and Supply of SCY-078
We have agreements with external vendors that are capable of supplying kilogram quantities of drug substance, and to produce drug product to support ongoing and planned clinical trials. However, we do not own or operate and do not intend to own or operate facilities for manufacturing, storage and distribution, or testing of drug substance or drug product. We have relied on third-party contract manufacturers for synthesis of our clinical compounds and manufacture of drug product. We expect to continue to rely on either existing or alternative third-party manufacturers to supply SCY-078 for ongoing and planned clinical trials and for commercial production.
SCY-078 is a semi-synthetic compound. Thus, the manufacturing process for SCY-078 involves fermentation and synthetic chemical steps. The process begins with fermentation to produce the natural product enfumafungin, which has been conducted by a third-party vendor on a scale sufficient to provide greater than 60kg of this starting material. Enfumafungin is then converted to SCY-078 via chemical modification in a series of chemical steps. Approximately 20 kg of drug substance has been manufactured. The overall process does not require any specialized equipment and uses readily sourced intermediates. At commercial launch, we expect cost of goods for SCY-078 to be similar to that of other small molecule drugs. We are negotiating agreements with large scale suppliers to produce both drug product and drug substance for our planned clinical trials. In the future, we plan to validate the process with selected vendors and secondary suppliers to establish a secure supply chain.
We estimate our supplies on hand for both oral and IV formulations to be sufficient to supply our ongoing Phase 1 and Phase 2 studies. Existing supplies would also support additional Phase 1 studies planned during the first half of 2016, including up to two drug-drug interaction studies with the oral formulation and multiple escalating dose studies for the IV formulation. Existing supplies have shown good stability to enable the completion of the clinical investigations as planned. Manufacture of additional supplies of SCY-078 drug substance is planned to support any further optimization of either of the formulations if needed. Additional batches of both oral and IV SCY-078 drug product will be manufactured as needed to support the subsequent stages of the clinical development plan.
A drug manufacturing program subject to extensive governmental regulations requires robust quality assurance systems and experienced personnel with the relevant technical and regulatory expertise as well as strong project management skills. We believe we have a team that is capable of managing these activities. The third-party vendors that currently manufacture clinical supplies to support our ongoing clinical studies have the necessary capabilities and are in compliance with cGMP appropriate for the current stage of development.
The third-party vendors we will select to support our manufacturing and supply program both for future late-stage development and commercial readiness activities will have the required capabilities with respect to facilities, equipment and technical expertise, quality systems that meet global regulatory and compliance requirements, satisfactory regulatory inspection history from relevant health authorities and proven track records in supplying drug substance and drug product for late-stage clinical and commercial use.
Our Cyclophilin Inhibitor Platform
We have developed a proprietary platform for cyclophilin inhibitors. Cyclophilins are a family of enzymes found in all mammalian cells which play a key role in a number of important cellular functions. Inhibiting cyclophilins shows promise as treatments for a range of diseases. To date, our cyclophilin inhibitor platform has produced two clinical stage compounds, described below.
SCY-635 is a novel, orally available cyclophilin inhibitor that has demonstrated clinical activity against HCV as a single agent and when dosed in combination with pegylated interferon and ribavirin. In these clinical studies, SCY-635 modified patients’ immune responses to HCV. These observations implicate cyclophilins in viral evasion of immune responses. HCV and Hepatitis B Virus are two of the most widespread global infections, with more than 170 million and 240 million chronic carriers respectively, and are leading causes of liver cirrhosis, liver cancer and liver transplantation. In October 2014, we granted Waterstone Pharmaceutical HK Limited, or "Waterstone", an international pharmaceutical business, exclusive worldwide rights to development and commercialization of SCY-635, and two additional compounds at Waterstone’s option, for the treatment of viral diseases in humans, under which we are entitled to receive potential milestones and royalties. Under the terms of our SCY-635 license agreement with Waterstone, we agreed that during the term of the agreement, we would not develop or commercialize, or grant any right or license to any third party to develop or commercialize, in Asia (excluding Japan), any cyclophilin inhibitor for treatment of viral diseases in humans.
SCY-641 is a novel cyclophilin inhibitor with activity similar to cyclosporine, the active ingredient in Restasis® and Optimmune®, products currently approved for dry eye disease in humans and dogs, respectively. The global human dry eye syndrome therapeutics market was valued at $1.8 billion in 2010 and the market value is expected to grow to $2.8 billion in 2017. Sales of Restasis® in 2012 were $792 million. SCY-641 has significantly improved water solubility compared to cyclosporine, which we believe will lead to improved tolerability and ease of use for treatment of dry eye disease (i.e., does not sting when applied and with anticipated required dosing of no more than twice daily). In August 2012, we licensed worldwide animal health rights for SCY-641 to Dechra Ltd., while retaining rights for human health indications. In November 2015, Dechra notified us of its intention to terminate its license agreement for the development of SCY-641 effective May 2016, returning full global rights of this compound to us. We are currently assessing the data generated by Dechra before deciding next steps and potentially identifying a new development and commercial partner for the human and animal health uses of SCY-641.
The key terms of our licensing agreements with Dechra and Waterstone are disclosed in the section below titled “Collaborations and Licensing Agreements.”
Our Contract Research and Development Services
As a spinout from Aventis in 2000, we began as a chemistry and animal health services company, providing contract research services to third parties through our Services Business. Through this Services Business, we built significant expertise in parasitic infections and drug discovery. Since our formation, we have expanded our animal health capabilities and have discovered a number of proprietary compounds. Through the provision of these contract research and development services, we built significant expertise in parasitic infections and drug discovery, including expanded animal health capabilities. The
Services Business generated substantially all of our revenues until we completed the sale of the Services Business to Accuratus Lab Services, Inc. in July 2015, as described further in the “Recent Developments” section of Item 7 of this Annual Report.
Research and Development Expenses
A significant portion of our operating expenses is related to research and development and we intend to maintain our strong commitment to research and development. In fiscal years 2015 and 2014 we spent $16.4 million and $8.3 million, respectively, on research and development expenses. See "Item 8. Financial Statements and Supplementary Data" of this Annual Report on Form 10-K for costs and expenses related to research and development, and other financial information for each of the fiscal years 2015 and 2014.
Collaborations and Licensing Agreements Associated with Our Core Drug Development Operations
We have a number of licensing and collaboration agreements associated with our core drug development operations, including the following:
Merck
We discovered and developed SCY-078 through a research collaboration with Merck Sharp & Dohme Corp., or "Merck", a subsidiary of Merck & Co., Inc. In May 2013 Merck transferred to us all development and commercialization rights for SCY-078 (also known as MK-3118). This decision was made following a review and prioritization of Merck’s infectious disease portfolio. Under the terms of the agreement, we received all human health rights to SCY-078, including all related technical documents, preclinical data, data from the seven Phase 1 trials conducted by Merck, and drug product and drug substance. The agreement continues until expiration of all royalty obligations. The agreement may be terminated if either party is in material breach and fails to remedy the breach after receiving written notice. In January 2014, Merck assigned the patents to us related to SCY-078 that it had exclusively licensed to us. Under the terms of the patent assignment, Merck no longer has responsibility to maintain the patents. Merck is eligible to receive milestones upon initiation of a Phase 3 clinical study, NDA filing and marketing approvals in each of the U.S., major European markets and Japan that could total up to $19 million. In addition, Merck will receive tiered royalties based on worldwide sales of SCY-078. The aggregate royalties are in the single digit percentages of net sales, and we expect to pay royalties on net sales of SCY-078 to Merck for no more than ten years from first commercial launch, on a country-by-country basis.
In December 2014, we entered into an amendment to the license agreement with Merck that defers the remittance of a milestone payment due to Merck, such that no amount will be due upon initiation of the first phase 2 clinical trial of a product containing the SCY-078 compound (the "Deferred Milestone"). The amendment also increased, in an amount equal to the Deferred Milestone, the milestone payment that will be due upon initiation of the first Phase 3 clinical trial of a product containing the SCY-078 compound. Except as described above, all other terms and provisions of the license agreement remain in full force and effect.
R-Pharm
In August 2013 we entered into an agreement with R-Pharm, CJSC, or "R-Pharm", a leading supplier of hospital drugs in Russia, granting them exclusive rights to develop and commercialize SCY-078 in the field of human health in Russia, Turkey, and certain Balkan, Central Asian, Middle Eastern and Northern African countries. We retained the right to commercialize SCY-078 in the Americas, Europe, and Asia. We received an upfront payment of $1.5 million and are entitled to receive up to $18 million in payments for development milestones and sales-based payments. We are also entitled to single digit percent royalty payments for products that do not fall under the patents and a royalty percentage in the teens for products that do fall under the patents. This agreement expires upon R-Pharm’s last royalty payment, which is the later of 12 years from the first registration of the product in the countries where R-Pharm’s license rights exist under this agreement, or the last to expire of the patents in such countries. Either party may terminate this agreement if the other party breaches, and fails to remedy the breach after receiving notice from the non-breaching party. We have the ability to terminate this agreement if we determine that R-Pharm fails to make reasonable progress in the development and commercialization of SCY-078. If we give R-Pharm notice of failure to make reasonable progress, R-Pharm will have the opportunity to correct the deficiencies.
The original agreement also included terms whereby R-Pharm would reimburse us for certain research and development costs associated with Phase 2 and Phase 3 clinical trials of oral SCY-078 and the development of an IV formulation of SCY-078. However, these cost reimbursement terms required that the clinical trials and the IV formulation development follow a global development plan that was agreed upon by both parties in August 2013. Subsequent to August 2013, modifications were made to the global development plan that caused the clinical trial cost reimbursement terms in the original agreement to no longer be enforceable. Further, the IV formulation development cost reimbursement terms in the original agreement did not specify which IV formulation and development costs were reimbursable by R-Pharm. In November 2014, we entered into a supplemental arrangement with R-Pharm, whereby R-Pharm was informed of the modified IV formulation development plan and R-Pharm agreed to reimburse us for specifically identified IV formulation development and manufacturing costs incurred
by us. The specifically identified costs were defined as all costs incurred by us under a separate arrangement we have with a third-party service provider, whereby the third-party service provider is performing certain IV formulation and development services for us. We estimate that total reimbursable costs pursuant to the original agreement and supplemental arrangement with R-Pharm will be approximately $1.3 to $1.9 million.
Dechra
In August 2012 we signed an agreement with Dechra Ltd., or "Dechra", a UK listed international veterinary pharmaceutical business, granting Dechra rights to SCY-641 for use in the field of animal health, including the treatment of canine keratoconjunctivitis sicca, or dry eye in dogs. Dechra was granted worldwide animal health rights and is responsible for the remaining clinical development and commercialization of SCY-641 in the animal health field. We retained the human health rights to the compound, including the right to use preclinical data generated by Dechra to support further human clinical development. Under the agreement, Dechra must use reasonable efforts to commercialize SCY-641. We received an upfront fee and are eligible to receive potential milestone payments up to £0.4 million as well as a royalty percentage in the low teens to the low twenties on the total net sales of product sales. Dechra’s obligations to pay royalties shall continue, on a product-by-product and country by country basis, until the later to expire of (i) all valid claims in such country and (ii) 12 years after the first commercial sale of such product in such country. This agreement expires when Dechra has completed all royalty payment obligations. If either party is in breach, and the breach continues after notice given by the non-breaching party, the non-breaching party may terminate the agreement. If we terminate the agreement because Dechra is in breach, Dechra must return all information required to be returned under the license agreement, free of charge, to us. If Dechra reasonably believes it is impossible to carry out further development or marketing of animal health products, Dechra may terminate this agreement at any time by giving us at least six months prior written notice. In November 2013, we amended this license agreement with Dechra in which we agreed to perform certain services for Dechra. In November 2015, Dechra notified us of its intention to terminate its license agreement for the development of SCY-641 effective May 2016, returning full global rights of this compound to us. We are currently assessing the data generated by Dechra before deciding next steps and potentially identifying a new development and commercial partner for the human and animal health uses of SCY-641.
Aventis
In May 2005, we entered into a license agreement with Aventis Pharma S.A., or "Aventis", a leading global healthcare company, pursuant to which Aventis granted us a worldwide license (with a right to sub-license) to certain of Aventis’s know-how, compounds and patents concerning cyclosporine derivatives exclusively in the field of treatment and prevention of HIV/AIDS and non-exclusively in all fields outside the treatment and prevention of HIV/AIDS. Under the terms of the agreement, we are obligated to maintain reasonable efforts to develop and commercialize a marketable product containing the subject compound and Aventis is responsible for maintaining and protecting the underlying patent rights. The agreement expires on a country by country basis at the end of the underlying intellectual property claims, and the expiration of the U.S. patent is December 23, 2017. We may terminate the agreement at any time, without cause, by giving Aventis 90 days notice. Aventis may terminate this agreement only if we commit a serious breach and fail to remedy the breach within 90 days of notice. Upon expiration of the agreement, we will have a fully paid-up, royalty free, worldwide, exclusive license in the field of treatment and prevention of HIV/AIDS and a non-exclusive license outside this field. We are obligated to pay Aventis up to an aggregate of $1.35 million in payments upon the achievement of certain milestones. In addition, on an annual basis, we will be obligated to pay a single digit percentage royalty on direct sales by us of all products developed under the agreement and we will pay a low single digit percentage of royalty on any sales by a sub-licensee of all products developed under the agreement. Pursuant to the terms of our October 2014 license agreement with Waterstone, Waterstone has agreed to reimburse us for any of these milestone payments that may become due to Aventis.
C-Chem
In June 2005, we entered into an assignment agreement with C-Chem AG, or "C-Chem", pursuant to which C-Chem assigned certain inventions, patents and know-how concerning cyclosporine derivatives for us to research, develop, manufacture and commercialize a product. Under the agreement, C-Chem has assigned to us all rights, title and interest in the subject patents as well as assigned all rights, title and interest to certain know-how with exclusive right to use and disclose the know-how for any purpose. Under the agreement, we must exercise reasonable commercial efforts to develop and commercialize a product using the licensed intellectual property and we are responsible for maintaining the licensed patents until the end of their lifetime. The U.S. patent on SCY-641 expires on June 10, 2019, and this agreement expires when no valid claim remains with respect to the underlying patents. C-Chem may terminate the agreement if an order by a court is made appointing a custodian, receiver, liquidator, assignee or trustee for us or if a court orders the winding up or liquidation of our affairs. We can terminate the agreement at any time by thirty (30) days written notice to C-Chem. If either party breaches any term or condition of the agreement, then the non-breaching party can terminate the agreement if notice is given to the breaching party and the breach is not remedied in sixty (60) days. Upon expiration of the agreement, we will have a fully paid-up, royalty free, worldwide exclusive license, and the right to grant sub-licenses, under the know-how and ancillary rights to commercialize and supply products. If the agreement is terminated by either party, we are obligated to reassign the patents, the
know-how and the ancillary rights to C-Chem, return any intellectual property to C-Chem, and cease all activities which would require a license under the subject patents. We paid C-Chem an initial payment of $0.3 million and a one-time $0.2 million milestone payment, and are obligated to pay C-Chem up to $0.95 million in payments upon the achievement of certain milestones. In addition, we will be obligated to pay a low single digit percentage royalty on direct sales by us of all products developed under the agreement and we will pay less than a 1% royalty on any sales by a licensee of all products developed under the agreement.
Waterstone
In October 2014, we entered into a license agreement with Waterstone, under which we granted Waterstone an exclusive, worldwide license to develop and commercialize SCY-635 for the treatment of viral diseases in humans. In addition, under the same agreement, we granted Waterstone an option for an exclusive, worldwide license to develop and commercialize two additional compounds of ours, SCY-575 and SCY-116, for the treatment of viral diseases in humans. The option is exercisable for a period of 18 months from the date of the agreement. In addition, we agreed that during the term of the agreement, we would not develop or commercialize, or grant any right or license to any third party to develop or commercialize, in Asia (excluding Japan), any cyclophilin inhibitor for treatment of viral diseases in humans.
The agreement expires upon Waterstone’s last royalty payment, which is the later of ten years from the last registration of the product, or the last to expire of the patents. Either party may terminate the agreement if the other party breaches and fails to remedy the breach after receiving notice from the nonbreaching party. Specifically, we have the ability to terminate the agreement if we determine that Waterstone failed to make reasonable progress in the development and commercialization of SCY-635 or the optioned compounds. If we give Waterstone notice of failure to make reasonable progress, Waterstone will have the opportunity to correct the deficiencies. If Waterstone fails to do so, we have the right to terminate the license.
We received an upfront license fee payment of $1.0 million in November 2014 for SCY-635, and we may receive an additional upfront payment of $0.5 million if Waterstone exercises its option for the two additional compounds. We are also entitled to receive certain payments on contingent future events, including 1) a development milestone payment of $4.0 million upon the first registration of a product, and 2) royalties based on a specified percentage of net sales (which percentage is in the mid-single digits), varying based on whether the product contains SCY-635 or one of the two additional compounds.
In January 2015, we entered into a patent assignment agreement with Waterstone under which a total of 18 patents and patent applications owned by us and relating to SCY-635 were assigned to Waterstone. Following this assignment, Waterstone assumed responsibility for maintenance, prosecution and enforcement of these patents and patent applications. We continue to remain the exclusive licensee of these Aventis Pharma patents, which continue to be exclusively sub-licensed to Waterstone, but were not assigned to Waterstone under the patent assignment agreement entered into in January 2015.
Collaborations and Licensing Agreements Associated with Our Former Services Business
We had a number of licensing and collaboration agreements associated with our former Services Business that were assigned to Accuratus in conjunction with the sale of the Services Business in July 2015, including the following:
Merial
Merial, a wholly owned subsidiary of Sanofi, is one of the largest animal health businesses in the world and had been a significant partner to our former Services Business since 2003. During 2015, we provided contract research and screening services in the field of animal health that primarily targeted parasites on a fee-for-service basis under an agreement effective December 2014. The Merial agreement was directly related to the Services Business and was assigned to Accuratus in conjunction with the sale of the Services Business in July 2015, including all future obligations and benefits. Revenue from the agreement is included in discontinued operations in the accompanying statements of operations.
In the year ended December 31, 2015, we received $2.1 million from Merial under the research services agreement, which accounted for 29% of our revenues in discontinued operations. No other customer, except for Elanco Animal Health, accounted for 10% or more of our revenues in discontinued operations during 2015.
Elanco Animal Health
In December, 2013, we entered into a license, development, and commercialization agreement with Elanco Animal Health, or "Elanco", the animal health division of Eli Lilly Company, an American global pharmaceutical company, pursuant to which we performed research services and granted to Elanco a worldwide license (with a right to sub-license) to certain of our know-how, compounds, and patents exclusively for applications and uses of parasiticides for animals (companion or food), animal products, animal feed, human food, or the food chain. The agreement with Elanco Licensing Agreement was directly related to the Services Business and was assigned to Accuratus in conjunction with the sale of the Services Business in July 2015, including all future obligations and benefits. Revenue from the agreement is included in discontinued operations in the
accompanying statements of operations. As of July 17, 2015, when the agreement was assigned to Accuratus, we had recognized revenues of $4.24 million under the agreement.
Government Regulation and Product Approval
Government regulation
Government authorities in the United States, at the federal, state and local level, and in other countries extensively regulate, among other things, the research, development, testing, manufacture, including any manufacturing changes, packaging, storage, recordkeeping, labeling, advertising, promotion, distribution, marketing, import and export of pharmaceutical products such as those we are developing. The processes for obtaining regulatory approvals in the United States and in foreign countries, along with subsequent compliance with applicable statutes and regulations, require the expenditure of substantial time and financial resources.
U.S. drug approval process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to a variety of administrative or judicial sanctions, such as the FDA’s refusal to approve pending NDAs, withdrawal of an approval, imposition of a clinical hold, issuance of warning letters, product recall requests, product seizures, total or partial suspension of production or distribution, injunctions, refusals of government contracts, restitution, disgorgement or civil or criminal penalties.
The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| |
• | completion of preclinical laboratory tests, animal studies and formulation studies in compliance with the FDA’s good laboratory practice, or GLP, regulations; |
| |
• | submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
| |
• | approval by an independent institutional review board, or IRB, at each clinical site before each trial may be initiated; |
| |
• | performance of adequate and well-controlled human clinical trials in accordance with good clinical practice, or GCP, to establish the safety and efficacy of the proposed drug for each indication, subject to on-going IRB review; |
| |
• | submission to the FDA of an NDA; |
| |
• | satisfactory completion of an FDA advisory committee review, if applicable; |
| |
• | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current Good manufacturing practice, or cGMP, regulations and guidance, and to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
| |
• | FDA review and approval of the NDA. |
Preclinical studies
Preclinical studies include laboratory evaluation of product chemistry, toxicity and formulation, as well as animal studies to assess potential safety and efficacy. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, among other things, to the FDA as part of an IND. Some preclinical testing may continue even after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions related to one or more proposed clinical trials and places the clinical trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. As a result, submission of an IND may not result in the FDA allowing clinical trials to commence.
Clinical trials
Clinical trials involve the administration of the investigational new drug to human subjects under the supervision of qualified investigators in accordance with GCP requirements, which include the requirement that all research subjects provide their informed consent in writing for their participation in any clinical trial. Clinical trials are conducted under protocols
detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated. A protocol for each clinical trial and any subsequent protocol amendments must be submitted to the FDA as part of the IND. In addition, an IRB at each institution participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that institution. Information about certain clinical trials must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their ClinicalTrials.gov website.
Human clinical trials are typically conducted in three sequential phases, which in some cases may overlap or be combined:
Phase 1: The drug is initially introduced into healthy human subjects or patients with the target disease or condition and tested for safety, dosage tolerance, absorption, metabolism, distribution, excretion and, if possible, to gain an early indication of its effectiveness.
Phase 2: The drug is administered to a limited patient population with the target disease to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage.
Phase 3: The drug is administered to an expanded patient population with the target disease, generally at geographically dispersed clinical trial sites, in well-controlled clinical trials to generate enough data to statistically evaluate the efficacy and safety of the product for approval, to establish the overall risk-benefit profile of the product, and to provide adequate information for the labeling of the product.
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently if serious adverse events occur. Phase 1, Phase 2 and Phase 3 clinical trials sometimes cannot be completed successfully within any specified period, or at all. Furthermore, the FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
In some cases, the FDA may condition approval of an NDA for a product candidate on the sponsor’s agreement to conduct additional clinical trials to further assess the drug’s safety and effectiveness after NDA approval. Such post-approval trials are typically referred to as Phase 4 studies.
In some circumstances, the FDA may also order a sponsor to conduct post-approval clinical trials if new safety information arises raising questions about the drug’s risk-benefit profile. Those clinical trials are typically referred to as Post-Marketing Requirements, or PMRs.
Marketing approval
Assuming successful completion of the required clinical testing, the results of the preclinical and clinical studies, together with detailed information relating to the product’s chemistry, manufacture, controls and proposed labeling, among other things, are submitted to the FDA as part of an NDA requesting approval to market the product for one or more indications. In most cases, the submission of an NDA is subject to a substantial application user fee. Under the Prescription Drug User Fee Act guidelines that are currently in effect, for a drug considered to be a new molecular entity, the FDA has a goal of twelve months from the date of the receipt of a standard non-priority NDA to review and act on the submission, or eight months for a priority NDA for such a new molecular entity drug.
In addition, under the Pediatric Research Equity Act of 2003, an NDA or supplement to an NDA must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements.
The FDA also may require submission of a risk evaluation and mitigation strategy, or REMS, to mitigate any identified or suspected serious risks. The REMS could include medication guides, physician communication plans, assessment plans, and elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission, before accepting them for filing, to determine whether they are sufficiently complete to permit substantive review. The FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The FDA reviews an NDA to determine, among other things, whether the drug is safe and effective and the facility in which it is manufactured, processed, packaged or held meets
standards designed to assure the product’s continued safety, quality and purity. The FDA is required to refer an application for a novel drug to an advisory committee or explain why such referral was not made. An advisory committee is a panel of independent experts, including clinicians and other scientific experts, that reviews, evaluates and provides a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
Before approving an NDA, the FDA typically will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving an NDA, the FDA will typically inspect one or more clinical sites to assure compliance with GCP.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The FDA may not grant approval on a timely basis, or at all.
If the FDA’s evaluation of the NDA and inspection of the manufacturing facilities are favorable, the FDA may issue an approval letter, or, in some cases, a complete response letter. A complete response letter generally contains a statement of specific conditions that must be met to secure final approval of the NDA and may require additional clinical or preclinical testing for the FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved indications for use for the product, require that contraindications, warnings or precautions be included in the product labeling, require that post-approval studies, including Phase 4 clinical trials, be conducted to further assess a drug’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution restrictions or other risk management mechanisms, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing studies or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes, and additional labeling claims, are subject to further testing requirements and FDA review and approval.
GAIN Act
The FDA has various programs, including fast track designation and priority review, that are intended to expedite or simplify the process for the development and FDA review of drugs that meet certain qualifications. The purpose of these programs is to provide important new drugs to patients earlier than under standard FDA review procedures.
The GAIN Act is intended to encourage development of new antibacterial and antifungal drugs for the treatment of serious or life-threatening infections by providing certain benefits to sponsors, including extended exclusivity periods, fast track and priority review. To be eligible for these benefits a product in development must seek and be awarded designation as a Qualifying Infectious Disease Product, or QIDP.
To qualify as a QIDP according to the criteria established in the GAIN Act a product must be an antibacterial or antifungal drug for human use intended to treat serious or life-threatening infections, including, those:
| |
(1) | caused by an antifungal resistant pathogen, including novel or emerging infectious pathogens; or |
| |
(2) | qualifying pathogens listed by the FDA in accordance with the GAIN Act. |
Fast Track Designation
The FDA is required to facilitate the development, and expedite the review, of drugs that are intended for the treatment of a serious or life-threatening disease or condition for which there is no effective treatment and which demonstrate the potential to address unmet medical needs for the condition. Under the Fast Track program, the sponsor of a new drug candidate may request that the FDA designate the drug candidate for a specific indication as a Fast Track drug concurrent with, or after, the filing of the IND for the drug candidate. The FDA must determine if the drug candidate qualifies for Fast Track designation within 60 days of receipt of the sponsor's request.
If a submission is granted Fast Track designation, the sponsor may engage in more frequent interactions with the FDA, and the FDA may review sections of the NDA before the application is complete. This rolling review is available if the applicant provides, and the FDA approves, a schedule for the submission of the remaining information and the applicant pays applicable user fees. However, the FDA's time period goal for reviewing an application does not begin until the last section of
the NDA is submitted. Additionally, Fast Track designation may be withdrawn by the FDA if the FDA believes that the designation is no longer supported by data emerging in the clinical trial process.
Post-approval requirements
Drugs manufactured or distributed pursuant to FDA approvals are subject to extensive and continuing regulation by the FDA, including, among other things, requirements relating to recordkeeping, periodic reporting, product sampling and distribution, advertising and promotion and reporting of adverse experiences with the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data.
The FDA may impose a number of post-approval requirements as a condition of approval of an NDA. For example, the FDA may require post-marketing testing, including Phase 4 clinical trials and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization.
In addition, drug manufacturers and other entities involved in the manufacture and distribution of approved drugs are required to register their establishments with the FDA and state agencies and are subject to periodic unannounced inspections by the FDA and these state agencies for compliance with cGMP requirements. Changes to the manufacturing process are strictly regulated and often require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting and documentation requirements upon the sponsor and any third-party manufacturers that the sponsor may decide to use. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance.
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
| |
• | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| |
• | warning letters or holds on post-approval clinical trials; |
| |
• | refusal of the FDA to approve pending NDAs or supplements to approved NDAs, or suspension or revocation of product license approvals; |
| |
• | product seizure or detention, or refusal to permit the import or export of products; or |
| |
• | injunctions or the imposition of civil or criminal penalties. |
The FDA strictly regulates marketing, labeling, advertising and promotion of products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability.
In addition, the distribution of prescription pharmaceutical products is subject to the Prescription Drug Marketing Act, or PDMA, and the Drug Supply Chain Security Act, or DSCSA, which regulate the distribution of drugs and drug samples at the federal level and set minimum standards for the registration and regulation of drug distributors by the states. Both the PDMA and state laws limit the distribution of prescription pharmaceutical product samples and, in conjunction with the DSCSA, impose requirements to ensure accountability in distribution.
Exclusivity and approval of competing products
Hatch-Waxman exclusivity
Market and data exclusivity provisions under the FDCA can delay the submission or the approval of certain applications for competing products, namely, abbreviated new drug applications, or ANDAs, for proposed generic drugs and 505(b)(2) NDAs for products that rely at least in part on FDA’s prior approval of another drug product. Drugs approved through a Section 505(b)(2) NDA generally include a clinically significant change from the previously approved drug product. Section 505(b)(2) permits the applicant to rely upon FDA’s approval of certain preclinical or clinical studies conducted for an approved product. The FDA typically requires companies to perform additional, sometimes extensive, clinical studies and analyses to support the change from the approved product. The FDA may then approve the new product candidate for all or some of the label
indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant.
The FDCA provides a five-year period of non-patent data exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active moiety. The active moiety of a drug is the molecule or ion responsible for the action of the drug substance, excluding any salts, esters or non-covalent derivatives that may be appended to the molecule or ion. During the exclusivity period for a new chemical entity, the FDA may not accept for review an ANDA or a 505(b)(2) NDA submitted by another company that references the previously approved drug. However, an ANDA or 505(b)(2) NDA may be submitted after four years if it contains a certification of patent invalidity or non-infringement.
The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA, or supplement to an existing NDA or 505(b)(2) NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant, are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages, strengths or dosage forms of an existing drug. This three-year exclusivity covers only the conditions of use associated with the new clinical investigations and, as a general matter, does not prohibit the FDA from approving ANDAs or 505(b)(2) NDAs for generic versions of the original, unmodified drug product. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
Pediatric exclusivity
Pediatric exclusivity is another type of non-patent marketing exclusivity in the United States and, if granted, provides for the attachment of an additional six months of regulatory protection to the term of any existing exclusivity, including the non-patent exclusivity periods described above, and to the regulatory term of any patent that has been submitted to FDA for the approved drug product. This six-month exclusivity may be granted based on the voluntary completion of a pediatric study or studies in accordance with an FDA-issued “Written Request” for such a study or studies.
Qualified Infectious Disease Product exclusivity
If the NDA for a QIDP is approved by the FDA, the FDA will extend by an additional five years any non-patent marketing exclusivity period awarded, such as a five-year exclusivity period awarded for a new chemical entity. This extension is in addition to any pediatric exclusivity extension awarded. Eligibility for the extension will be denied if the product is approved for uses that would not meet the definition of a QIDP.
Foreign regulation
To market any product outside of the United States, we would need to comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy and governing, among other things, clinical trials, marketing authorization, commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we would need to obtain the necessary approvals by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others.
Pharmaceutical coverage, pricing and reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any drug products for which we may obtain regulatory approval. Sales of any of our product candidates which may be ultimately approved, including SCY-078, will depend, in part, on the extent to which the costs of the products will be covered by third-party payors, including government health programs such as Medicare and Medicaid, and commercial health insurers. The process for determining whether a payor will provide coverage for a drug product is separate from the process for determining the reimbursement rate for the drug product once coverage is approved. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the approved drugs for a particular indication or may apply utilization management requirements such as prior authorization to restrict access to certain approved drugs for a particular indication.
To secure coverage and reimbursement for any product that might be approved by the FDA for sale, we may need to conduct expensive pharmacoeconomic studies to demonstrate the medical necessity and cost-effectiveness of the product, in addition to the costs required to obtain FDA or other comparable regulatory approvals. Our product candidates may not be considered medically necessary or cost-effective by government or private third-party payor decision makers. A payor’s
decision to provide coverage for a drug product does not mean that the product will be adequately reimbursed. Third-party reimbursement may not be sufficient to enable us to realize an appropriate return on our investment in product development.
The containment of healthcare costs has become a priority of federal, state and foreign governments, and the prices of drugs have been a focus in this effort. Third-party payors are increasingly challenging the prices of medical products and corresponding services and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. If these third-party payors do not consider products for which we may obtain regulatory approval to be cost-effective compared to other available therapies, they may not provide coverage for our products as a benefit under their health insurance plans or, if they do, the reimbursement rates may not be adequate to allow recovery of product development and production costs.
The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs to limit the growth of government-paid health care costs, including price controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs. Adoption of such controls and measures, and tightening of restrictive policies in jurisdictions with existing controls and measures, could limit payments for pharmaceuticals such as the drug product candidates that we are developing and could adversely affect our net revenue and results.
Pricing and reimbursement requirements vary widely from country to country. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of a particular product candidate to currently available therapies.
The marketability and adoption of any products for which we receive regulatory approval for commercial sale may suffer if the government and private third-party payors fail to provide adequate coverage and reimbursement. In addition, emphasis on managed care in the United States has increased, and we expect will continue to increase, the pressure on drug pricing. Coverage policies, third-party payment rates and drug pricing regulation may change at any time. In particular, in March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010, together the Affordable Care Act, was adopted in the United States. This law substantially changes the way healthcare is financed by both governmental and private insurers, and significantly impacts the pharmaceutical industry. Changes that may affect our business if we or our partners commercialize our products in the future include those governing enrollment in federal healthcare programs, reimbursement changes, rules regarding prescription drug benefits under the health insurance exchanges, and fraud and abuse and enforcement.
Healthcare law and regulation
Healthcare providers, physicians and third-party payors often play a primary role in the recommendation and prescription of any product candidates for which we may obtain marketing approval. Our future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute our products for which we obtain marketing approval. Restrictions under applicable federal and state healthcare laws and regulations include the following:
| |
• | The federal healthcare anti-kickback statute prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made, in whole or in part, under federally funded healthcare programs such as Medicare and Medicaid. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the federal anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal healthcare programs. The Affordable Care Act clarified that a person or entity need not have actual knowledge of the federal anti-kickback statute or specific intent to violate it. In addition, the Affordable Care Act amended the federal civil False Claims Act to provide that a claim that includes items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. There are a number of statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, however, the exceptions and safe harbors are drawn narrowly, and practices that do not fit squarely within an exception or safe harbor may be subject to scrutiny. |
| |
• | The federal civil False Claims Act imposes civil penalties, and provides for whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, claims for payment of government funds that are false or fraudulent or knowingly making, or causing to be made, a false record or statement material to a false or fraudulent claim to avoid, decrease or conceal an obligation to pay money to the federal government. Several pharmaceutical and other healthcare companies have faced enforcement actions |
under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, federal anti-kickback statute violations and certain marketing practices, including off-label promotion, may also implicate the federal civil False Claims Act. Federal civil False Claims Act violations may result in civil monetary damages and penalties and exclusion from participation in federal healthcare programs. There are also criminal penalties, including imprisonment and criminal fines, for making or presenting a false, fictitious or fraudulent claim to the federal government.
| |
• | The federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act, imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program and also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information. |
| |
• | The federal criminal false statements statute prohibits knowingly and willfully falsifying, concealing or covering up a material fact, making any materially false, fictitious, or fraudulent statement or representation, or making or using any false writing or document knowing the same to contain any materially false, fictitious or fraudulent statement or entry in connection with the delivery of or payment for healthcare benefits, items or services. |
| |
• | The federal Physician Payment Sunshine Act, being implemented as the Open Payments Program, requires applicable pharmaceutical manufacturers of covered drugs to engage in extensive tracking of physician and teaching hospital payments, maintenance of a payments database, and public reporting of the payment data. Pharmaceutical manufacturers with products for which payment is available under Medicare, Medicaid or the State Children’s Health Insurance Program must perform such tracking and provide annual reports through the Open Payments Program. CMS posts such manufacturer disclosures on a searchable public website. Failure to comply with the reporting obligations may result in civil monetary penalties. |
| |
• | Analogous state laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by Medicaid or other state programs or, in several states, apply regardless of the payor. Several state laws require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products in those states and to report gifts and payments to individual health care providers in those states. Some of these states also prohibit certain marketing related activities including the provision of gifts, meals or other items to certain health care providers. In addition, California, Connecticut, Nevada, and Massachusetts require pharmaceutical companies to implement compliance programs or marketing codes. |
Intellectual Property
We strive to protect the proprietary technology that we believe is important to our business, including seeking and maintaining patents intended to cover our product candidates and compositions, their methods of use and processes for their manufacture and other inventions that are commercially important to the development of our business. We also rely on trade secrets to protect aspects of our business that are not amenable to, or that we do not consider appropriate for, patent protection.
As of March 1, 2016, we are the owner of 12 issued U.S. patents and 102 issued non-U.S. patents with claims to novel compounds, compositions containing them, processes for their preparation, their uses as pharmaceutical agents and test methods, with terms expiring between 2017 and 2029. Of these patents, one U.S. patent relates to SCY-078. We are actively pursuing three U.S. patent applications (provisional and non-provisional), two international (PCT) patent applications and 39 non-U.S. patent applications in at least 28 jurisdictions.
We are the exclusive licensee from Aventis Pharma of two issued U.S. patents and eight issued non-U.S. patents, with claims to novel compounds, compositions containing them, processes for their preparation, and their uses as pharmaceutical agents, with terms expiring between 2017 and 2019. These include patents covering our lead cyclophilin inhibitor compound, SCY-635, which, in October 2014 was exclusively licensed to Waterstone for the treatment of viral diseases in humans (as described in the section above titled “Collaborations and Licensing Arrangements”). The agreement granted Waterstone an exclusive license or sub-license for human viral diseases to certain patents and patent applications owned by or exclusively licensed to us relating to SCY‑635. In January 2015, we entered into a patent assignment agreement with Waterstone under which a total of 18 patents and patent applications owned by us and relating to SCY-635 were assigned to Waterstone. Following this assignment, Waterstone assumed responsibility for maintenance, prosecution and enforcement of these patents and patent applications. We continue to remain the exclusive licensee of these Aventis Pharma patents, which continue to be
exclusively sub-licensed to Waterstone, but were not assigned to Waterstone under the patent assignment agreement entered into in January 2015.
Our success will depend significantly on our ability to obtain and maintain patents and other proprietary protection for commercially important technology, inventions and know-how related to our business, defend and enforce our patents, maintain our licenses to use intellectual property owned by third parties, preserve the confidentiality of our trade secrets and operate without infringing the valid and enforceable patents and other proprietary rights of third parties. We also rely on know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen, and maintain our proprietary position in the field of antifungal agents.
We believe that we have a strong intellectual property position and substantial know-how relating to the development and commercialization of SCY-078, including patents or patent applications covering inventions that we have co-invented with Merck. We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications filed by us in the future, nor can we be sure that any of our existing patents or any patents that may be granted to us in the future will be commercially useful in protecting our technology.
Our objective is to continue to expand our intellectual property estate by filing patent applications directed to SCY-078 or derivatives thereof. We intend to pursue, maintain, and defend patent rights, whether developed internally or licensed from third parties, and to protect the technology, inventions, and improvements that are commercially important to the development of our business.
SCY-078
The patent portfolio for SCY-078 is directed to cover compositions of matter, formulation, methods of use and precursors or intermediaries in its preparation. This patent portfolio includes an issued U.S. patent and corresponding foreign national and regional counterpart patents and patent applications. The patents and patent applications relating to SCY-078 include patents and patent applications that were initially assigned to us and Merck Sharp & Dohme Corp, a subsidiary of Merck & Co., Inc. Merck Sharp & Dohme Corp. subsequently assigned to us all of its rights in these patents and patent applications relating to SCY-078. The issued composition of matter patent (U.S. Patent No. 8,188,085), if the appropriate maintenance, renewal, annuity, and other governmental fees are paid, is expected to expire in 2030. Based on our current development plan, we believe that an additional term of up to five years for the SCY-078 U.S. patent may result from the patent term extension provision of the Drug Price Competition and Patent Term Restoration Act of 1984 (the Hatch-Waxman Act). We expect that the patent applications in this portfolio, if issued, and if appropriate maintenance, renewal, annuity, and other governmental fees are paid, would expire between 2029 and 2035, including any additional term from patent term adjustment or patent term extension. The patent term calculation method and the provisions under the Hatch-Waxman Act are described in the “Patent Term” section below. We are not currently aware of any third-party patents (other than patents we have licensed) encompassing SCY-078.
The terms of issued SCY-078 composition of matter patents in other jurisdictions (Algeria, Armenia, Australia, Azerbaijan, Belarus, China, Colombia, Europe, Honduras, Hong Kong, Indonesia, Japan, Lebanon, Kazakhstan, Kyrgyzstan, Mexico, Moldova, Morocco, New Zealand, Nicaragua, Peru, Philippines, Russia, Singapore, South Africa, Taiwan, Tajikistan, Tunisia, Turkmenistan and Ukraine), if the appropriate maintenance, renewal, annuity, and other government fees are paid, are expected to expire in 2029. These patents and patent applications (if applicable), depending on the national laws, may benefit from extension of patent term in individual countries. In some European countries, for example, a supplementary protection certificate, if obtained, provides a maximum of five years of market exclusivity. The duration of the supplementary protection certificate may be extended to five and a half years when the supplementary protection certificate relates to a human medicinal product for which data from clinical trials conducted in accordance with an agreed Pediatric Investigation Plan, or PIP, have been submitted. Likewise, in Japan, the term of a patent may be extended by a maximum of five years in certain circumstances.
SCY-641
The patent portfolio for SCY-641 is directed to cover compositions of matter, formulation, and methods of use. This patent portfolio includes issued U.S. patents and corresponding foreign national and regional counterpart patents and patent applications. The patents and patent applications relating to SCY-641 include patents and patent applications owned by us. The issued composition of matter patent (U.S. Patent No. 6,583,265), if the appropriate maintenance, renewal, annuity, and other government fees are paid, is expected to expire in 2019. The issued methods of use patents (U.S. Patent Nos. 8,188,052 and 8,551,952), if the appropriate maintenance, renewal, annuity, and other government fees are paid, are expected to expire in 2029 or 2027, respectively. We believe that the term for up to five years for one of the SCY-641 U.S. patents may be extended under the patent term extension provision of the Hatch-Waxman Act. We expect that the patent applications in this portfolio, if issued, and if appropriate maintenance, renewal, annuity, and other governmental fees are paid, would expire between 2019 and 2034, including any additional term from patent term adjustment or patent term extension, assuming that five year extension is granted. The patent term calculation method and the provisions under the Hatch-Waxman Act are described in the “Patent Term” section below.
The term of issued SCY-641 composition of matter patents in other jurisdictions (Australia, Canada, China, Europe and Japan) and methods of use patents and patent applications (if applicable) relating to SCY-641 (in Australia, Canada, China, Europe, Japan and South Africa), if the appropriate maintenance, renewal, annuity, and other government fees are paid, are expected to expire between 2019 and 2027. The patents and patent applications (if applicable), covering SCY-641, depending on the national laws, may also benefit from extension of patent term in individual countries.
Other product candidates
In addition to SCY-078, SCY-635 and SCY-641, we have a chemical library of more than 1,000 macrocyclic compounds generated by the research team at SCYNEXIS. This library includes compounds which are covered by patents or patent applications filed by us, but also includes novel chemical compounds which could form the basis for future patent applications.
Patent Term
The term of individual patents and patent applications will depend upon the legal term of the patents in the countries in which they are obtained. Generally, the patent term is 20 years from the date of filing of the patent application (or earliest filed parent application, if applicable).
Under the Hatch-Waxman Act, the term of a patent that claims an FDA-approved drug may also be eligible for patent term extension, or PTE. Eligibility for a PTE is based, in part, on whether the FDA approval of the drug represents the first permitted commercial marketing or use of the drug. Drugs that are considered to be new chemical entities under FDA’s regulations are generally eligible for PTE.
PTE permits patent term restoration of a U.S. patent as partial compensation for patent term lost during the FDA regulatory review process, which includes both the testing period while the drug is being investigated under an IND and the approval period while FDA is reviewing a marketing application. The length of the patent term extension is half the testing period plus all of the approval period, with certain limitations. The Hatch-Waxman Act permits a patent term extension of up to five years beyond the expiration of the patent; however, a patent term extension cannot in any event extend the remaining term of a patent beyond a total of 14 years from the date of product approval; only one patent that claims an approved drug may be extended; and the applicable approval must be the first approval of the product under the provision of law authorizing the approval. During the extension period, the patent holder’s rights under the patent are generally limited to approved uses of the product. Similar provisions may be available in Europe and certain other foreign jurisdictions to extend the term of a patent that covers an approved drug. When possible, depending upon the length of clinical trials and other factors involved in the filing of an NDA, we expect to apply for patent term extensions for patents covering SCY-078 and its use in treating various diseases. As a specific example, if we are awarded the maximum length of PTE, our U.S. composition of matter patent relating to SCY-078 would have an expected expiration date of the earlier of 14 years from product approval or August 28, 2035. However, depending on any changes in our clinical path and the date of FDA approval, the PTE may not be granted, or may be less than the maximum.
Proprietary rights and processes
We may rely, in some circumstances, on proprietary technology and processes (including trade secrets) to protect our technology. However, these can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by entering into confidentiality agreements with our employees, consultants, scientific advisors, contractors, and collaborators. We also seek to preserve the integrity and confidentiality of our proprietary technology and processes by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our proprietary technology and processes may otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants, scientific advisors, contractors, or collaborators use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions. For this and more comprehensive risks related to our proprietary technology and processes, please see the section on “Risk Factors—Risks Relating to Our Intellectual Property.”
Legal Proceedings
From time to time, we are involved in various legal proceedings arising in the ordinary course of our business. We are not currently a party to any legal proceedings the outcome of which, if determined adversely to us, would individually or in the aggregate have a material effect on our business, operating results, financial condition or cash flows.
Employees
As of March 1, 2016, we had 11 employees, all of whom were employed on a full-time basis. Our employees are engaged in administration, accounting and finance, research, clinical development, manufacturing, and business development functions. We believe our relations with our employees are good.
Corporate Information
We were incorporated in the State of Delaware on November 4, 1999. Our corporate headquarters are located at 101 Hudson Street, Suite 3610, Jersey City, New Jersey 07302.
Our corporate website address is www.scynexis.com. Our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act are available free of charge on our website. The information contained on, or that can be accessed through, our website is not part of this Annual Report, and the inclusion of our website address in this Annual Report is an inactive textual reference only.
Facilities
Our corporate headquarters are located in Jersey City, New Jersey in a subleased office space consisting of approximately 10,141 square feet. The sublease for this facility expires in July 2018.
In evaluating our business, you should carefully consider the following risks, as well as the other information contained in this Annual Report on Form 10-K. These risk factors could cause our actual results to differ materially from those contained in forward-looking statements we have made in this Annual Report on Form 10-K and those we may make from time to time. If any of the following risks actually occurs, our business, financial condition and operating results could be harmed. The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties not presently known to us, or that we currently see as immaterial, may also harm our business.
Risks Relating to Our Financial Condition and Need for Additional Capital
We have never been profitable, we have no products approved for commercial sale, and to date we have not generated any revenue from product sales. As a result, our ability to curtail our losses and reach profitability is unproven, and we may never achieve or sustain profitability.
We are not profitable and do not expect to be profitable in the foreseeable future. We have incurred net losses in each year since our inception, including a net loss of approximately $32.6 million for the year ended December 31, 2015, and expect to incur a net loss for the year ending December 31, 2016. As of December 31, 2015, we had an accumulated deficit of approximately $150.1 million. Although we have generated revenue through our Services Business prior to the sale of the Services Business in July 2015, these revenues historically have not been sufficient to support our business, and so in addition we have financed our operations through the sale of convertible preferred stock, convertible debt, and common stock. On a prospective basis, our strategic focus, along with the commitment of our financial resources, will be directed towards the development of SCY-078, our lead product candidate. We have not generated any revenue from product sales. Although we had cash and cash equivalents of $47.0 million as of December 31, 2015, there can be no assurances that we will be able to continue our operations on a long-term basis. We have suffered substantial losses from operations since inception and will require additional financing.
We expect to continue to incur significant expenses and increasing operating losses for the foreseeable future. The net losses we incur may fluctuate significantly from quarter to quarter. We anticipate that our expenses will increase substantially as we:
| |
• | continue the development of SCY-078 for treatment of multiple indications; |
| |
• | conduct ongoing and initiate new clinical trials for SCY-078; |
| |
• | seek marketing approvals for SCY-078; |
| |
• | establish a sales, marketing and distribution infrastructure to commercialize any products for which we may obtain marketing approval; |
| |
• | maintain, expand and protect our intellectual property portfolio; |
| |
• | hire additional clinical, quality control and scientific personnel; |
| |
• | maintain and create additional infrastructure to support our operations as a public company; and |
| |
• | develop in-house product candidates or seek to in-license product candidates from third-parties. |
In addition, our expenses could increase if we are required by the U.S. Food and Drug Administration, or the FDA, to perform studies in addition to, or that are larger than, those that we currently expect.
As a result of the foregoing, we expect to experience net losses and negative cash flows from operations for the foreseeable future, and we are unable to predict when, or if, we will be able to achieve profitability. Our losses and negative cash flows have had, and will continue to have, an adverse effect on our stockholders’ equity, financial position and working capital.
We expect a number of factors to cause our operating results to fluctuate on a quarterly and annual basis, which may make it difficult to predict our future performance.
Our financial condition and operating results have varied significantly in the past and will continue to fluctuate from quarter to quarter or year to year due to a variety of factors, many of which are beyond our control. The following factors relating to our business, as well as factors described elsewhere in this report, may contribute to these fluctuations:
| |
• | the costs associated with developing SCY-078, which are difficult for us to predict; |
| |
• | any delays in regulatory review and approval of SCY-078; |
| |
• | delays in the timing of submission of a new drug application, or NDA, as well as commencement, enrollment and the timing of clinical testing, of SCY-078 or any other product candidates we may seek to develop; |
| |
• | our ability to commercialize product candidates, both in the United States and overseas, if we are able to obtain regulatory approval to do so; |
| |
• | the costs associated with obtaining and maintaining regulatory approval and ongoing company compliance and product compliance for SCY-078; |
| |
• | market acceptance of SCY-078 and any future product candidates we may seek to develop; |
| |
• | changes in regulations and regulatory policies; |
| |
• | competition from existing products or new products that may emerge; |
| |
• | the ability of patients or healthcare providers to obtain coverage of, or sufficient reimbursement for, any products we are able to develop; |
| |
• | our ability to establish or maintain collaborations, licensing or other arrangements; |
| |
• | costs related to, and outcomes of, potential litigation; |
| |
• | potential product liability claims; and |
| |
• | potential liabilities associated with hazardous materials. |
Due to the various factors mentioned above, and others, the results of any quarterly or annual periods should not be relied upon as indications of future operating performance.
We may continue to require substantial additional capital, and if we are unable to raise capital when needed we would be forced to delay, reduce or eliminate our development program for SCY-078.
Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is expensive. If the FDA requires that we perform additional studies beyond those that we currently expect, our expenses could increase beyond what we currently anticipate, the timing of the submission of the NDA could be delayed, and any potential product approval could be delayed. We believe that our existing cash and cash equivalents as of December 31, 2015, will be sufficient to meet our anticipated operating requirements into the second quarter of 2017; provided, however, that changing circumstances may cause us to consume cash more rapidly than we currently anticipate. We may need to raise additional funds from additional issuances of equity and/or debt securities or otherwise obtain funding through strategic alliances or collaborations with third parties. In any event, we will require additional capital to complete development of, to seek regulatory approval for and, if approval is obtained, to commercialize SCY-078 and any future product candidates we may seek to develop. Raising funds in the current economic environment, when the capital markets have been affected by the global recession, may present additional challenges.
When we are required to secure additional financing, the additional fundraising efforts may divert our management from our day-to-day activities, which may adversely affect our ability to develop and commercialize SCY-078 and any future product
candidates we may seek to develop. In addition, we cannot guarantee that financing will be available in sufficient amounts or on terms acceptable to us, if at all. If we are unable to raise additional capital when required or on acceptable terms, we may be required to:
| |
• | significantly delay, scale back or discontinue the development or commercialization of SCY-078 and any future product candidates we may seek to develop; |
| |
• | seek strategic alliances for research and development programs at an earlier stage than otherwise would be desirable or on terms that are less favorable than might otherwise be available; or |
| |
• | relinquish or license on unfavorable terms our rights to any product candidates that we otherwise would seek to develop or commercialize ourselves. |
If we are required to conduct additional fundraising activities and we are unable to raise additional capital in sufficient amounts or on terms acceptable to us, we will be prevented from pursuing development and commercialization efforts, which will have a material adverse effect on our business, operating results and prospects.
We have a significant concentration of credit risk in the form of cash on deposit with two banks, which exceeds the individual account FDIC insurance limits.
We had cash and cash equivalents of $47.0 million on deposit with two banking institutions as of December 31, 2015. We monitor the credit rating of our commercial banks based on the quarterly reviews of independent analysts. If the commercial banks experience insolvency and we are unable to access our cash and cash equivalents, or if we experience a loss of principal, it may adversely affect our ability to develop and commercialize SCY-078 and any future product candidates we may seek to develop.
Risks Relating to the Development, Regulatory Approval and Commercialization of Our Product Candidates For Human Use
Historically we have been primarily a contract research and development services company devoting a majority of our resources and efforts to providing research and development services to other companies, and we only recently shifted our primary focus to developing our own drug candidate, SCY-078.
We were spun out from Aventis in 2000 as a chemistry and animal health services company, providing contract research services to third parties. Since then, until our disposition of this business in July 2015, we have derived substantially all of our revenue from providing these services to human and animal health companies to assist them in developing their own drug candidates. In the course of providing these services, we leveraged this expertise to develop our own proprietary compounds, including a platform of cyclophilin inhibitors, among them SCY-635, which we exclusively licensed to Waterstone in October 2014 in the field of human health.
Although we have conducted Phase 1 and Phase 2 studies of SCY-635, our cyclophilin inhibitor that we exclusively licensed to Waterstone in October 2014 in the field of human health, we only acquired the rights to develop SCY-078, our lead drug candidate for the treatment of invasive fungal infections, in May 2013. We do not have a significant history of developing our own drug candidates, and we have not brought any drug candidates to market, which makes it difficult to assess our ability to develop and commercialize SCY-078 and any future product candidates we may seek to develop or commercialize.
We cannot be certain that SCY-078 will receive regulatory approval, and without regulatory approval we will not be able to market SCY-078. Regulatory approval is a lengthy, expensive and uncertain process.
Our ability to generate significant revenue related to SCY-078 sales will depend on the successful development and regulatory approval of SCY-078. We expect that the earliest that we could obtain regulatory approval of SCY-078 and commence commercialization of SCY-078 will be several years from now, if at all.
We currently have no products approved for sale and we cannot guarantee that we will ever have marketable products. The development and commercialization of a product candidate, including preclinical and clinical testing, manufacturing, quality systems, labeling, approval, record-keeping, selling, promotion, marketing and distribution of products, is subject to extensive regulation by the FDA in the United States and regulatory authorities in other countries, with regulations differing from country to country. We are not permitted to market product candidates in the United States until and unless we receive approval of an NDA from the FDA. We have not submitted an NDA for SCY-078. Obtaining approval of an NDA is a lengthy, expensive and uncertain process. An NDA must include extensive preclinical and clinical data and supporting information to establish the product candidate’s safety and effectiveness for each indication. The approval application must also include
significant information regarding the chemistry, manufacturing and controls for the product. The product development and regulatory review process typically takes years to complete, involves numerous uncertainties and the potential for concerns to emerge late in the development process, and approval is never guaranteed. Even if a product is approved, the FDA may limit the indications for which the product may be used, include extensive warnings on the product labeling or require costly ongoing requirements for post-marketing clinical studies and surveillance or other risk management measures to monitor the safety or efficacy of the product candidate, including the imposition of a Risk Evaluation and Mitigation Strategy, or REMS. Markets outside of the United States also have requirements for approval of drug candidates with which we must comply prior to marketing. Obtaining regulatory approval for marketing of a product candidate in one country does not ensure we will be able to obtain regulatory approval in other countries, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in other countries. Also, any regulatory approval of a product candidate, once obtained, may be withdrawn. If SCY-078 or any of our other wholly-owned or partnered product candidates do not receive timely regulatory approval, or fail to maintain that regulatory approval, we may not be able to generate sufficient revenue to become profitable or to continue our operations. Moreover, the filing of our NDA or the receipt of regulatory approval does not assure commercial success of any approved product.
Although both the oral and IV formulations of SCY-078 have been granted Qualified Infectious Disease Product status and Fast Track designation, this does not guarantee that the length of the FDA review process will be significantly shorter than otherwise, or that SCY-078 will ultimately be approved by the FDA.
We applied to the FDA for, and received, the designation of the oral tablet and the IV formulations of SCY-078 as Qualified Infectious Disease Product, or QIDP, under the Generating Antibiotic Incentives Now Act, or GAIN Act. We also applied to the FDA for, and were granted, Fast Track designation. However, the primary framework of the GAIN Act became effective July 9, 2012, and as a relatively new law there is limited precedent for the way in which it will be implemented. Receipt of QIDP status and Fast Track designation in practice may not result in a faster development process, review or approval compared to drugs considered for approval under conventional FDA procedures and does not assure ultimate approval by the FDA or related exclusivity benefits.
Delays in the commencement, enrollment and completion of clinical trials could result in increased costs to us and delay or limit our ability to obtain regulatory approval for SCY-078 or any future product candidates.
We do not know whether clinical trials of SCY-078 or any future product candidates we may seek to develop will be allowed to commence or, if commenced, will be completed on schedule or at all. The commencement, enrollment and completion of clinical trials can be delayed for a variety of reasons, including:
| |
• | inability to reach agreements on acceptable terms with prospective clinical research organizations, or CROs, and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| |
• | difficulty identifying and engaging qualified clinical investigators; |
| |
• | regulatory objections to commencing a clinical trial or proceeding to the next phase of investigation, including inability to reach agreement with the FDA or non-U.S. regulators regarding the scope or design of our clinical trials or for other reasons such as safety concerns that might be identified during preclinical development or early stage clinical trials; |
| |
• | inability to identify and maintain a sufficient number of eligible trial sites, many of which may already be engaged in other clinical trial programs, including some that may be for the same indication as our product candidates; |
| |
• | withdrawal of clinical trial sites from our clinical trials as a result of changing standards of care; |
| |
• | inability to obtain institutional review board (or ethics review committee) approval to conduct a clinical trial at prospective sites; |
| |
• | difficulty identifying, recruiting and enrolling eligible patients to participate in clinical trials for a variety of reasons, including meeting the enrollment criteria for our study and competition from other clinical trial programs for the same indication as product candidates we seek to commercialize; |
| |
• | inability to retain patients in clinical trials due to the treatment protocol, personal issues, side effects from the therapy or lack of efficacy; |
| |
• | inability to produce and/or obtain in a timely manner sufficient quantity of our products to satisfy the requirements of the clinical trials; and |
| |
• | inability to obtain sufficient funding to commence a clinical trial. |
In addition, a clinical trial may be suspended or terminated by us, our current or any future partners, an institutional review board, the FDA or other regulatory authorities due to a number of factors, including:
| |
• | failure by us, CROs or clinical investigators to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| |
• | failed inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities; |
| |
• | unforeseen safety or efficacy issues or any determination that a clinical trial presents unacceptable health risks; or |
| |
• | lack of adequate funding to continue the clinical trial due to unforeseen costs resulting from enrollment delays, requirements to conduct additional trials and studies, increased expenses associated with the services of our CROs and other third parties, or other reasons. |
If we are required to conduct additional clinical trials or other testing of SCY-078 or any future product candidates we may seek to develop, we may be delayed in obtaining, or may not be able to obtain, marketing approval for these product candidates.
In addition, if our current or any future partners have rights to and responsibility for development of SCY-078 or any future product candidates, they may fail to meet their obligations to develop and commercialize the product candidates, including clinical trials for these product candidates.
Changes in regulatory requirements and guidance may occur and we or any of our partners may be required by appropriate regulatory authorities to amend clinical trial protocols to reflect these changes. Amendments may require us or any of our partners to resubmit clinical trial protocols to independent review boards for re-examination, which may impact the costs, timing or successful completion of a clinical trial. If we or any of our partners experience delays in the completion of, or if we or our partners terminate, clinical trials, the commercial prospects for SCY-078 and any future product candidates we may seek to develop will be harmed, and our ability to generate revenue from sales of these product candidates will be prevented or delayed. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of a product candidate.
Delays in the patient enrollment in our Phase 2 Invasive Candidiasis trial, including delays associated with the implementation of recent protocol amendments, potential additional protocol amendments that we are currently evaluating, and the opening of additional investigational sites inside and outside the US, could have an adverse effect on the costs and timing of our SCY-078 development efforts.
Our multicenter Phase 2 study for invasive candidiasis with primary endpoints of safety, tolerability, and pharmacokinetics of the oral formulation of SCY-078 as step-down treatment in patients initially treated with echinocandin therapy for invasive Candida infections is ongoing. We have opened new investigational sites in the U.S. and in Latin America and we are in the process of opening more sites in these regions and in Europe. Based on the data collected on the enrolled patients, together with the data from our recently completed Phase 1 biocomparison study, we expect to achieve the primary objectives of the study with fewer patients than originally planned and to report top line data by the end of the second quarter of 2016. If these actions are not successful and we are unable to enroll a sufficient number of patients, it may delay and/or adversely affect the data we expect to receive from the study, limit our ability to achieve the study's primary objectives, or cause us to terminate the study before it is completed.
Clinical failure can occur at any stage of clinical development. Because the results of earlier clinical trials are not necessarily predictive of future results, any product candidate we or our current or potential future partners advance through clinical trials may not have favorable results in later clinical trials or receive regulatory approval.
Clinical failure can occur at any stage of clinical development. Clinical trials may produce negative or inconclusive results, and we or our partners may decide, or regulators may require us, to conduct additional clinical or preclinical testing. In addition, data obtained from tests are susceptible to varying interpretations, and regulators may not interpret data as favorably as we do, which may delay, limit or prevent regulatory approval. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will generate the same results or otherwise provide adequate data to demonstrate the efficacy and safety of a product candidate. Frequently, product candidates that have shown promising results in early clinical trials have subsequently suffered significant setbacks in later clinical trials. In addition, the design of a clinical trial can determine whether its results will support approval of a product application, or approval of a supplemental application to add a new indication or other changes, and flaws or shortcomings in the design of a clinical trial may not become apparent until the clinical trial is well advanced. We have limited experience in designing clinical trials and may be unable to design and execute a clinical trial to
support regulatory approval, or approval of supplemental applications for new indications or other changes. Further, clinical trials of potential products often reveal that it is not practical or feasible to continue development efforts. If SCY-078 or any future product candidates are found to be unsafe or lack efficacy, we or our collaborators will not be able to obtain regulatory approval for them and our business would be harmed. For example, if the results of our ongoing or planned Phase 2 and Phase 3 clinical trials of SCY-078 do not achieve, to the satisfaction of regulators, the primary efficacy endpoints and demonstrate an acceptable level of safety, the prospects for approval of SCY-078 would be materially and adversely affected. A number of companies in the pharmaceutical industry, including those with greater resources and experience than us, have suffered significant setbacks in Phase 2 and Phase 3 clinical trials, even after seeing promising results in earlier clinical trials.
In some instances, there can be significant variability in safety and/or efficacy results between different trials of the same product candidate due to numerous factors, including differences in trial protocols and design, differences in size and type of the patient populations, adherence to the dosing regimen and the rate of dropout among clinical trial participants. Further, the patients taking SCY-078 often have other significant medical issues, such as organ transplants, cancer or other conditions in which their immune systems are suppressed, which makes it difficult to measure the effect of SCY-078 in the presence of these medical issues. We do not know whether any Phase 2, Phase 3 or other clinical trials we or any partners may conduct will demonstrate consistent and/or adequate efficacy and safety to obtain regulatory approval to market SCY-078 and any future product candidates we may seek to develop.
We have limited experience in conducting clinical trials and have never submitted an NDA before, and we may be unable to do so for SCY-078 or any future product candidate we may seek to develop.
Merck completed seven Phase 1 clinical trials of SCY-078, we have completed one Phase 1 clinical trial, and have initiated one additional Phase 1 and two Phase 2 trials, which are ongoing. We are planning to conduct additional Phase 1, Phase 2, and Phase 3 clinical trials of SCY-078. The conduct of successful Phase 2 and Phase 3 clinical trials is essential in obtaining regulatory approval, and the submission of a successful NDA is a complicated process. We have limited experience in preparing and submitting regulatory filings, have previously only sponsored one Phase 2 clinical trial, and have not previously sponsored any Phase 3 clinical trials, nor have we ever submitted an NDA. Consequently, we may be unable to successfully and efficiently execute and complete these planned clinical trials in a way that is acceptable to the FDA and leads to an NDA submission, acceptance and approval of SCY-078 or any future product candidate we may seek to develop. We may require more time and incur greater costs than our competitors and may not succeed in obtaining regulatory approvals of product candidates that we may seek to develop. In addition, failure to commence or complete, or delays in, our planned clinical trials would prevent us from or delay us in commercializing SCY-078 or any future product candidate we may develop.
The environment in which our regulatory submissions may be reviewed changes over time, which may make it more difficult to obtain regulatory approval of any of our product candidates we may seek to develop or commercialize.
The environment in which regulatory submissions are reviewed changes over time. For example, average review times at the FDA for NDAs have fluctuated over the last ten years, and we cannot predict the review time for any submission with any regulatory authorities. Review times can be affected by a variety of factors, including budget and funding levels and statutory, regulatory and policy changes. Moreover, in light of widely publicized events concerning the safety risks of certain drug products, regulatory authorities, members of Congress, the Government Accountability Office, medical professionals and the general public have raised concerns about potential drug safety issues. These events have resulted in the withdrawal of drug products, revisions to drug labeling that further limit use of the drug products and establishment of risk evaluation and mitigation strategies that may, for instance, restrict distribution of drug products. The increased attention to drug safety issues may result in a more cautious approach by the FDA to clinical trials. Data from preclinical studies and clinical trials may receive greater scrutiny with respect to safety, which may make the FDA or other regulatory authorities more likely to terminate clinical trials before completion, or require longer or additional clinical trials that may result in substantial additional expense, a delay or failure in obtaining approval or approval for a more limited indication or conditions of use than originally sought.
In addition, data obtained from preclinical studies and clinical trials are subject to different interpretations, which could delay, limit or prevent regulatory review or approval of product candidates. Changes in FDA personnel responsible for review of our submissions could also impact the manner in which our data are viewed. Furthermore, regulatory attitudes towards the data and results required to demonstrate safety and efficacy can change over time and can be affected by many factors, such as the emergence of new information, including information on other products, policy changes and agency funding, staffing and leadership. We do not know whether future changes to the regulatory environment will be favorable or unfavorable to our business prospects.
If SCY-078 or any other future product candidates for which we receive regulatory approval do not achieve broad market acceptance, the revenue that is generated from their sales will be limited.
The commercial success of SCY-078 or any other product candidates we may seek to develop will depend upon the acceptance of these product candidates among physicians, patients, the medical community and healthcare payors. The degree of market acceptance of product candidates will depend on a number of factors, including:
| |
• | limitations or warnings contained in the FDA-approved labeling; |
| |
• | changes in the standard of care for the targeted indications; |
| |
• | limitations in the approved indications; |
| |
• | availability of alternative therapies with potentially advantageous results, or other products with similar results at similar or lower cost, including generics and over-the-counter products; |
| |
• | lower demonstrated clinical safety or efficacy compared to other products; |
| |
• | occurrence of significant adverse side effects; |
| |
• | ineffective sales, marketing and distribution support; |
| |
• | lack of availability of reimbursement from managed care plans and other third-party payors; |
| |
• | timing of market introduction and perceived effectiveness of competitive products; |
| |
• | lack of cost-effectiveness; |
| |
• | adverse publicity about our product candidates or favorable publicity about competitive products; |
| |
• | lack of convenience and ease of administration; and |
| |
• | potential product liability claims. |
If SCY-078 or any future product candidates we may seek to develop are approved, but do not achieve an adequate level of acceptance by physicians, healthcare payors and patients, sufficient revenue may not be generated from these product candidates, and we may not become or remain profitable. In addition, efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may never be successful.
A significant use of antifungal drugs consists of treatment due to the presence of symptoms before diagnosis of the invasive fungal infections, and if recently approved diagnostic tools, or additional tools currently under development, for the quick diagnosis of invasive fungal infections are broadly used in the marketplace, the number of treatments using antifungal drugs may decrease significantly, decreasing the potential market for SCY-078.
We believe that a large portion of the treatments using antifungal drugs are administered when symptoms of invasive fungal infections are present but a diagnosis of the infection has not yet been made, due to the rapid and potentially fatal progression of invasive fungal infections. Diagnostic tools recently approved by the FDA, or currently under development, for the rapid diagnosis of invasive fungal infections may significantly diminish the need to treat patients in advance of diagnosis of invasive fungal infections, which will reduce the potential market for SCY-078 in the event that we are able to obtain FDA approval of SCY-078. Moreover, if a rapid and accurate test of the susceptibility of a fungal infection to generically available treatments is developed and widely adopted, the market for SCY-078 may suffer.
If resistance to SCY-078 develops quickly or cross-resistance with echinocandins becomes more common, our business will be harmed.
We recognize that, over time, resistance develops against every antibacterial and antifungal drug. One or more strains of fungal pathogens may develop resistance to SCY-078 more rapidly than we currently expect, either because our hypothesis of the mechanism of action is incorrect or because a strain of fungi undergoes some unforeseen genetic mutation that permits it to survive. Since we expect lower resistance relative to other antifungal drug classes to be a major factor in the commercialization of SCY-078, rapid development of such resistance or development of cross resistance with echinocandins would have a major adverse impact on the acceptability and sales of SCY-078.
If we are unable to obtain regulatory approval of both the oral and IV formulations of SCY-078, SCY-078 may not achieve broad market acceptance and sales will be limited.
Current treatment regimens for invasive fungal infections typically involve initial administration of treatments as an IV infusion, with a switch to an oral formulation of the same or a similar medication to complete the course of treatment on an out-patient basis. We believe that providing both the IV and oral formulations will be beneficial to doctors who prefer to start treatment of patients in a hospital setting with an IV therapy and then switch them to an oral formulation of the same medication. If we are unable to successfully develop and achieve regulatory approval for either the oral or IV formulation of SCY-078, our lead product candidate may not achieve, or may be delayed in achieving, broad market acceptance and sales will be limited.
Our product candidates may have undesirable side effects that may delay or prevent marketing approval, or, if approval is received, require them to be taken off the market or otherwise limit their sales.
It is impossible to predict when or if SCY-078 or any other product candidate we may seek to develop will prove effective or safe or will receive marketing approval. Unforeseen side effects from any product candidates could arise either during clinical development or, if approved, after the product has been marketed. For example, the most frequently noted adverse effects reported as associated with SCY-078 treatment in the Phase 1 studies of SCY-078 conducted to date were diarrhea, abdominal pain, headache, nausea, fatigue, increased orthostatic heart rate, abnormal GI sounds, vomiting and dizziness. Serious adverse events are common when conducting clinical trials in seriously ill population such as patients experiencing invasive candidiasis. Several serious adverse events have been reported in our clinical trials, but only one has been deemed to be related to the study drug. This event was in a subject who experienced significant liver function test increases after receiving a single dose of orally administered SCY-078; the subject recovered without any intervention. Another subject also reported elevation of liver function test after receiving oral SCY-078; this case also fully recovered without intervention and was not deemed serious by the investigator. Preclinical findings in the future could trigger the need to evaluate or monitor for specific potential safety concerns in clinical trials. The results of future clinical trials may show that SCY-078 and any future product candidates we may seek to develop cause undesirable or unacceptable side effects, which could interrupt, delay or halt clinical trials, resulting in delay of, or failure to obtain, marketing approval from the FDA and other regulatory authorities, or may lead us to abandon their development altogether.
Even if SCY-078 or any future product candidate we may seek to develop receives marketing approval, we or others may subsequently identify undesirable or unacceptable side effects caused by these products, in which case:
| |
• | regulatory authorities may require the addition of labeling statements, specific warnings, precautions, contraindications or field alerts to physicians and pharmacies; |
| |
• | we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| |
• | we may have limitations on how we promote the product; |
| |
• | sales of the product may decrease significantly; |
| |
• | regulatory authorities may require us to take our approved product off the market; |
| |
• | we may be subject to litigation or product liability claims; and |
| |
• | our reputation may suffer. |
Any of these events could prevent us or our current or potential future partners from achieving or maintaining market acceptance of the affected product or could substantially increase commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenue from the sale of products.
We have never marketed a drug before, and if we are unable to establish an effective sales force and marketing infrastructure or enter into acceptable third-party sales and marketing or licensing arrangements, we may not be able to successfully commercialize SCY-078 and any future product candidates we may seek to develop.
We currently do not have any sales, distribution and marketing capabilities, the development of which will require substantial resources and will be time consuming. The costs incurred in the development of these capabilities, either internally or through a third-party contract sales organization, would be incurred in advance of any approval of a product candidate. In addition, we may not be able to hire a sales force in the United States that is sufficient in size or has adequate expertise in the
medical markets that we intend to target. If we are unable to establish our sales force and marketing capability, our operating results may be adversely affected. In addition, we plan to enter into sales and marketing or licensing arrangements with third parties for international sales of any approved products. If we are unable to enter into or maintain any such arrangements on acceptable terms, or at all, we may be unable to market and sell SCY-078 or any future product candidates we may seek to develop in these markets.
We expect that SCY-078 and any future product candidates we may seek to develop will face competition, and most of our competitors have significantly greater resources than we do.
The pharmaceutical industry is highly competitive, with a number of established, large pharmaceutical companies, as well as many smaller companies. There are many foreign and domestic pharmaceutical companies, biotechnology companies, public and private universities, government agencies and research organizations actively engaged in research and development of products that may target the same markets as SCY-078 and any future product candidates we may seek to develop. We expect any products we develop to compete on the basis of, among other things, product efficacy, price, lack of significant adverse side effects and convenience and ease of treatment. For example, SCY-078 will compete against current leading antifungal drugs, including voriconazole from the azole class, caspofungin from the echinocandin class, and liposomal amphotericin B from the polyenes class, many of which are currently available in generic form, or expected to be available in generic form at the time SCY-078 might be approved.
Compared to us, many of our competitors in the antifungal market have, and potential competitors for any future product candidates we may seek to develop may have, substantially greater:
| |
• | resources, including capital, personnel and technology; |
| |
• | research and development capability; |
| |
• | clinical trial expertise; |
| |
• | intellectual property portfolios; |
| |
• | expertise in prosecution of intellectual property rights; |
| |
• | manufacturing and distribution expertise; and |
| |
• | sales and marketing expertise. |
As a result of these factors, our competitors and potential competitors may obtain regulatory approval of their products more rapidly than we do. Our competitors and potential competitors may also develop drugs that are more effective, more widely used and less costly than ours and may also be more successful than us in manufacturing and marketing their products and maintaining compliance with ongoing regulatory requirements.
Reimbursement decisions by third-party payors may have an adverse effect on pricing and market acceptance in the United States. If there is not sufficient reimbursement for our products, it is less likely that our products will be purchased by patients and/or providers.
Successful commercialization of pharmaceutical products usually depends on the availability of adequate coverage and reimbursement from third-party payors, including commercial insurers and federal and state healthcare programs. Patients and/or healthcare providers who purchase drugs generally rely on third-party payors to reimburse all or part of the costs associated with such products. As such, adequate coverage and reimbursement from third-party payors can be essential to new product acceptance and may have an effect on pricing.
Because SCY-078 is not currently commercially available, we do not know the extent to which it will be reimbursed if it is approved by the FDA. If we choose to bring other product candidates to market, they will be subject to similar uncertainty. We believe that SCY-078 and any other product candidates that are brought to market are less likely to be purchased by patients and/or providers if they are not adequately reimbursed by third-party payors.
Furthermore, the market for our product candidates may depend on access to third-party payors’ drug formularies, or lists of medications for which third-party payors provide coverage and reimbursement. Industry competition to be included in such formularies results in downward pricing pressures on pharmaceutical companies. Third-party payors may refuse to include a particular branded drug in their formularies when a competing generic product is available. The adoption of certain payment methodologies by third-party payors may limit our ability to profit from the sale of SCY-078. For example, under Medicare,
hospitals are reimbursed under an inpatient prospective payment system. This pricing methodology provides a single payment amount to hospitals based on a given diagnosis-related group. As a result, with respect to Medicare reimbursement for services in the hospital inpatient setting, hospitals could have a financial incentive to use the least expensive drugs for the treatment of invasive fungal infections, particularly the IV formulations of these drugs, as they are typically administered in the hospital, which may significantly impact our ability to charge a premium for SCY-078.
All third-party payors, whether governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs, including mechanisms to encourage the use of generic drugs. Congress has also considered policies to lower the reimbursement formulas in federal and state healthcare programs. Furthermore, coverage of, and reimbursement for, drugs can differ significantly from payor to payor and may require significant time and resources to obtain. In addition, new laws or regulations could impact future coverage and reimbursement.
Healthcare policy changes, including the Affordable Care Act, may have a material adverse effect on us.
In recent years, there have been numerous initiatives on the federal and state levels for comprehensive reforms affecting the payment for, the availability of and reimbursement for healthcare services in the United States, including pharmaceutical products. These initiatives have ranged from proposals to fundamentally change federal and state healthcare reimbursement programs, including providing comprehensive healthcare coverage to the public under governmental funded programs, to minor modifications to existing programs.
In March 2010, Congress enacted the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or the Affordable Care Act. The Affordable Care Act is designed to expand access to affordable health insurance, control healthcare spending, and improve healthcare quality. The law includes provisions to tie Medicare provider reimbursement to healthcare quality and incentives, mandatory compliance programs, enhanced transparency disclosure requirements, increased funding and initiatives to address fraud and abuse, and incentives to state Medicaid programs to expand their coverage and services. It also imposes an annual tax on pharmaceutical manufacturers or importers who sell “branded prescription drugs.” Implementation of the Affordable Care Act is occurring on an ongoing basis, and it is unclear what effect the Affordable Care Act or other state proposals may have on our business.
In addition to the Affordable Care Act, there will continue to be proposals by legislators at both the federal and state levels, regulators and third-party payors to keep drug costs down. Certain of these changes could impose limitations on the prices we will be able to charge for any products that are approved or the amounts of reimbursement available for these products from governmental agencies or third-party payors or may increase the tax requirements for life sciences companies such as ours. We anticipate that the Affordable Care Act and other future healthcare reform proposals could have a material adverse effect on our industry, and may limit our ability to commercialize SCY-078 and any future product candidates we may seek to develop and/or invest in new development.
We expect that a portion of the market for SCY-078 and any other product candidates we may seek to develop will be outside the United States. However, our product candidates may never receive approval or be commercialized outside of the United States.
Before we or any commercial partners can market and commercialize any product candidates outside of the United States, there are numerous and varying regulatory requirements of other countries that will apply. Research and marketing authorization procedures vary among countries and can involve additional product testing and administrative review periods. The marketing authorization process in other countries may include all of the risks detailed above regarding failure to obtain FDA approval in the United States as well as other risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country, or identification of potential safety concerns in one country, may have a negative effect on the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval could have the same adverse effects detailed above regarding FDA approval in the United States. As described above, such effects include the risks that:
| |
• | SCY-078 and any future product candidates we may seek to develop may not generate preclinical or clinical data that are deemed sufficient by regulators in a given jurisdiction; |
| |
• | SCY-078 may not be approved for all indications requested, or any indications at all, in a given jurisdiction which could limit the uses of SCY-078 and any future product candidates we may seek to develop and have an adverse effect on product sales and potential royalties; and |
| |
• | such approval in a given jurisdiction may be subject to limitations on the indicated uses for which the product may be marketed or require costly post-marketing follow-up studies. |
Foreign countries may have requirements for marketing authorization holders or distributors to have a legal or physical presence in that country, and consideration of and compliance with these requirements may result in additional time and expense before we can pursue or obtain marketing authorization in foreign jurisdictions. If we do receive approval in other countries, we may enter into sales and marketing arrangements with third parties for international sales of any approved products.
Even if SCY-078 or any other future product candidates we may seek to develop receive regulatory approval, we may still face future development and regulatory difficulties.
Even if regulatory approval is obtained for SCY-078 or any other future product candidates we may seek to develop, regulatory authorities may still impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies. Given the number of high profile adverse events with certain drug products, regulatory authorities may require, as a condition of approval, costly risk evaluation and mitigation strategies, which may include safety surveillance, restricted distribution and use, patient education, enhanced labeling, expedited reporting of certain adverse events, pre-approval of promotional materials and restrictions on direct-to-consumer advertising. For example, any labeling approved for any of our product candidates may include a restriction on the term of its use, or it may not include one or more intended indications. Furthermore, any new legislation addressing drug safety issues could result in delays or increased costs during the period of product development, clinical trials and regulatory review and approval, as well as increased costs to assure compliance with any new post-approval regulatory requirements. Any of these restrictions or requirements could force us or our partners to conduct costly studies.
SCY-078 and any other future product candidates we may seek to develop will also be subject to ongoing regulatory requirements for the packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information on the drug. In addition, approved products, manufacturers and manufacturers’ facilities are required to comply with extensive FDA requirements, including ensuring that quality control and manufacturing procedures conform to current Good Manufacturing Practices, or cGMP. As such, we and our contract manufacturers, which we will be responsible for overseeing and monitoring for compliance, are subject to continual review and periodic inspections to assess compliance with cGMP. Accordingly, we and others with whom we work must continue to expend time, money and effort in all areas of regulatory compliance, including manufacturing, production and quality control. The FDA may hold us responsible for any deficiencies or noncompliance of our contract manufacturers in relation to SCY-078 and any other future product candidates we may seek to develop. Failure to follow cGMP can result in products being deemed adulterated, which carries significant legal implications. We will also be required to engage in pharmacovigilance activities and report certain adverse reactions and production problems, if any, to the FDA and to comply with certain requirements concerning advertising and promotion for products. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved label. As such, we may not promote products for indications or uses for which they do not have approval. Failure to comply with FDA advertising and promotion standards, which are often subject to interpretation by regulators, may result in a wide range of exposure and liability for us.
If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured or disagrees with the promotion, marketing or labeling of a product, a regulatory agency may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If the manufacturing or marketing of products fail to comply with applicable regulatory requirements, a regulatory agency may:
| |
• | mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners; |
| |
• | require us or our partners to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
| |
• | impose other civil or criminal penalties; |
| |
• | suspend regulatory approval; |
| |
• | suspend any ongoing clinical trials; |
| |
• | refuse to approve pending applications or supplements to approved applications filed by us, our partners or our potential future partners; |
| |
• | impose restrictions on operations, including costly new manufacturing requirements; or |
| |
• | seize or detain products or require a product recall. |
Non-compliance may also open a company to potential whistleblower lawsuits and the potential for liability under the False Claims Act.
Pharmaceutical companies are subject to significant ongoing regulatory obligations and oversight, which may result in significant additional expense and limit our ability to commercialize our products.
We are subject to regulation by other regional, national, state and local agencies, including the Department of Justice, the Office of Inspector General of the U.S. Department of Health and Human Services and other regulatory bodies. Violations of any of the foregoing requirements could result in penalties being assessed against us.
The federal Anti-Kickback Statute prohibits, among other things, knowingly and willfully offering, paying, soliciting, or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical companies on one hand and prescribers, purchasers and formulary managers on the other. The Affordable Care Act, among other things, clarified that a person or entity need not have actual knowledge of the federal Anti-Kickback Statute or specific intent to violate it. In addition, the Affordable Care Act amended the federal civil False Claims Act to provide that a claim that includes items or services resulting from a violation of the federal anti-kickback statute, constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. There are a number of statutory exceptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, however, the exceptions and safe harbors are drawn narrowly, and practices that do not fit squarely within an exception or safe harbor may be subject to scrutiny.
The federal civil False Claims Act prohibits any person from knowingly presenting, or causing to be presented, claims for payment of government funds that are false or fraudulent or knowingly making, or causing to be made, a false record or statement material to a false or fraudulent claim to avoid, decrease or conceal an obligation to pay money to the federal government. Many pharmaceutical and other healthcare companies have been investigated and have reached substantial financial settlements with the federal government under these laws for a variety of alleged marketing activities, including providing free product to customers with the expectation that the customers would bill federal programs for the product; providing consulting fees, grants, free travel, and other benefits to physicians to induce them to prescribe the company’s products; and inflating prices reported to private price publication services, which are used to set drug payment rates under government healthcare programs. Companies have been prosecuted for causing false claims to be submitted because of the marketing of their products for unapproved uses. Pharmaceutical and other healthcare companies have also been prosecuted on other legal theories of Medicare and Medicaid fraud.
The majority of states also have statutes or regulations similar to the federal Anti-Kickback Statute and federal civil False Claims Act, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Some of these states also prohibit certain marketing related activities including the provision of gifts, meals, or other items to certain health care providers. In addition, certain states, including California, Connecticut, Nevada, and Massachusetts, require pharmaceutical companies to implement compliance programs or marketing codes.
Compliance with various federal and state laws is difficult and time consuming, and companies that violate them may face substantial penalties. The potential sanctions include civil monetary penalties, exclusion of a company’s products from reimbursement under government programs, criminal fines and imprisonment. Because of the breadth of these laws and the lack of extensive legal guidance in the form of regulations or court decisions, it is possible that some of our business activities or those of our commercial partners could be subject to challenge under one or more of these laws. Such a challenge could have a material adverse effect on our business and financial condition and growth prospects.
We could become subject to government investigations and related subpoenas. Such subpoenas are often associated with previously filed qui tam actions, or lawsuits filed under seal under the federal civil False Claims Act. Qui tam actions are brought by private plaintiffs suing on behalf of the federal government for alleged federal civil False Claims Act violations. The time and expense associated with responding to such subpoenas, and any related qui tam or other actions may be extensive, and we cannot predict the results of our review of the responsive documents and underlying facts or the results of such actions. Responding to government investigations, defending any claims raised, and any resulting fines, restitution, damages and penalties, settlement payments or administrative actions, as well as any related actions brought by stockholders or other third parties, could have a material impact on our reputation, business and financial condition and divert the attention of our management from operating our business.
The number and complexity of both federal and state laws continues to increase, and additional governmental resources are being added to enforce these laws and to prosecute companies and individuals who are believed to be violating them. In
particular, the Affordable Care Act includes a number of provisions aimed at strengthening the government’s ability to pursue federal Anti-Kickback Statute and federal False Claims Act cases against pharmaceutical manufacturers and other healthcare entities, including substantially increased funding for healthcare fraud enforcement activities, enhanced investigative powers, and amendments to the federal False Claims Act that make it easier for the government and whistleblowers to pursue cases for alleged kickback and false claim violations. Responding to a government investigation or enforcement action would be expensive and time-consuming and could have a material adverse effect on our business and financial condition and growth prospects.
If we fail to comply with applicable federal, state, or local regulatory requirements, we could be subject to a range of regulatory actions that could affect our ability to commercialize our products and could harm or prevent sales of any affected products that we are able to commercialize, or could substantially increase the costs and expenses of commercializing and marketing our products. Any threatened or actual government enforcement action could also generate adverse publicity and require that we devote substantial resources that could otherwise be used in other aspects of our business.
Regulations, guidelines and recommendations published by various government agencies and organizations may affect the use of SCY-078 and any future product candidates we may seek to develop.
Government agencies may issue regulations and guidelines directly applicable to us, our partners or our potential future partners and our product candidates. In addition, professional societies, practice management groups, private health/science foundations and organizations involved in various diseases from time to time publish guidelines or recommendations to the healthcare and patient communities. These various sorts of recommendations may relate to such matters as product usage, dosage, and route of administration and use of related or competing therapies. Changes to these recommendations or other guidelines advocating alternative therapies could result in decreased use of SCY-078 and any future product candidates we may seek to develop, which may adversely affect our results of operations.
Risks Relating to Our Drug Development Activities and Former Contract Research and Development Services
As a result of the divestiture of our former contract research and development business, we now contract with a third-party provider for certain drug development activities related to SCY-078, and if these services are terminated or are not as effective as when we could provide them internally, our development of SCY-078 may be delayed or harmed.
In connection with the sale of our former contract research and development business (“Services Business”) to Accuratus, we entered into the Services Agreement (“Services Agreement”) with Accuratus pursuant to which Accuratus will provide us with certain contract research and development services for 18 months following the closing of the sale of the Services Business. The purpose of the Services Agreement is to replace drug development services for the advancement of SCY-078 that were previously provided internally by our employees prior to the sale of the Services Business. These former employees have extensive knowledge and expertise pertaining to our SCY-078 drug development activities, and we are substantially dependent upon the continued access to their expertise pursuant to the terms of the Services Agreement. If we lose our ability to access this expertise, we could experience a significant delay in both identifying another comparable provider and then contracting for its services, which could adversely affect our development efforts. We may be unable to retain an alternative provider on reasonable terms, or at all. Even if we locate an alternative provider, it is likely that any provider will need additional time to respond to our needs and may not provide the same or similar type or level of services, which could have an adverse affect on the cost and timing of our development activities related to SCY-078.
We face potential liability and exposure as a result of the prior performance of our contract research and development services, and if successful claims are brought against us, we may incur substantial liability, which may exceed the revenues we have received for the prior performance of our contract research and development services.
To date substantially all of our revenue has been generated from the former provision of our contract research and development services. In the event that a regulator asserts that we have conducted activities in a non-compliant manner or a former customer asserts that we have conducted our contract research and development services negligently, or otherwise asserts that as a result of the performance of our contract research and development services for that client we have somehow harmed their business or the prospects of their product candidates, we could be subject to litigation, which could divert management’s attention from the operation of our business, including the development of SCY-078, or subject us to indemnification obligations to Accuratus under the terms of our Agreement with them in connection with the divestiture of our Services Business. Further, if such litigation is successful, or if we determine that we must settle the litigation, we could be forced to pay substantial damages, which could be more than the revenues that we generated from that customer, as the services that we performed are only a small portion of the development efforts of our customers. Even if we are successful in defending any such claims, we could incur substantial legal costs to do so. Further, publicity of any such litigation or claims could hurt
our reputation. Any such litigation against us could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
Risks Related to Our Dependence on Third Parties
We are dependent on our existing third-party collaboration with R-Pharm to commercialize SCY-078 in the Russian Federation and certain other countries, and if R-Pharm is not successful in commercializing SCY-078 in those countries, we will lose a significant source of potential revenue.
We currently have a development license and supply agreement with R-Pharm, CJSC, or R-Pharm, a leading supplier of hospital drugs in Russia, pursuant to which we license to R-Pharm rights to develop and commercialize SCY-078 in the field of human health in Russia and certain smaller non-core markets. R-Pharm will pay us milestone payments upon the achievement of specified milestones, including registration of SCY-078 in a country and upon the achievement of specified levels of sales. In addition, R-Pharm will pay us royalties upon sales of SCY-078 by R-Pharm. We are relying on R-Pharm to commercialize SCY-078 in the countries covered by our agreement with it, and if R-Pharm is not able to commercialize SCY-078 in those countries, or determines not to pursue commercialization of SCY-078 in those countries, we will not receive any milestone or royalty payments under the agreement.
We are dependent on other third-party collaborations to develop and commercialize product candidates we have outlicensed, and if our third-party collaborators are not successful in developing and commercializing product candidates we have outlicensed, we will not receive any revenue from these collaborations.
A portion of our strategy is to license to third parties rights to develop and commercialize product candidates, including candidates we have discovered other than SCY-078, and if these third parties do not perform under our agreements with them, we will not receive any revenue from these collaborations. For example, we currently have license agreements with R-Pharm, CSJC, or R-Pharm, to develop and commercialize SCY-078 in Russia and several smaller non-core markets and with Waterstone Pharmaceutical, or Waterstone, to develop and commercialize SCY-635 for the treatment of viral diseases in humans. We are relying on R-Pharma and Waterstone to commercialize the compounds subject to the respective license agreements, and if either is not able to commercialize the compounds subject to the respective agreements, or determines not to pursue commercialization of the compounds, we will not receive any royalty payments under the agreements. We are also party to a license agreement with Dechra Ltd, or Dechra, pursuant to which we licensed to Dechra rights to develop and commercialize SCY-641 for use in animal health, and we were eligible to receive royalties on sales of products developed with SCY-641. In November 2015, we received a notice of termination from Dechra indicating it would terminate the license agreement for SCY-641 effective in May 2016. If our third-party collaborators under the R-Pharma and Waterstone agreements and any future agreements we enter into do not perform under the agreements, or terminate the agreements, we will not receive the benefits we expect under the agreements.
We may not be successful in establishing and maintaining development and commercialization collaborations, which could adversely affect our ability to develop and commercialize product candidates.
Developing pharmaceutical products, conducting clinical trials, obtaining regulatory approval, establishing manufacturing capabilities and marketing approved products is expensive. Consequently, we plan to establish collaborations for development and commercialization of product candidates and research programs. For example, we currently have a development license and supply agreement with R-Pharm, pursuant to which we license to R-Pharm rights to develop and commercialize SCY-078 in the field of human health in Russia and certain smaller non-core markets, and if SCY-078 receives marketing approval, we may enter into additional sales and marketing arrangements with third parties for international sales. If we are unable to enter into any of these arrangements on acceptable terms, or at all, we may be unable to market and sell SCY-078 and any future product candidates we may seek to develop in certain markets. We expect to face competition in seeking appropriate collaborators. Moreover, collaboration arrangements are complex and time consuming to negotiate, document and implement and they may require substantial resources to maintain. We may not be successful in our efforts to establish and implement collaborations or other alternative arrangements for the development of product candidates. When we partner with a third party for development and commercialization of a product candidate, we can expect to relinquish to the third party some or all of the control over the future success of that product candidate. Our collaboration partner may not devote sufficient resources to the commercialization of product candidates or may otherwise fail in their commercialization. The terms of any collaboration or other arrangement that we establish may not be favorable to us. In addition, any collaboration that we enter into may be unsuccessful in the development and commercialization of product candidates. In some cases, we may be responsible for continuing preclinical and initial clinical development of a partnered product candidate or research program, and the payment we receive from our collaboration partner may be insufficient to cover the cost of this development.
If we are unable to reach agreements with suitable collaborators for product candidates, we could face increased costs, we may be forced to limit the number of product candidates we can commercially develop or the territories in which we commercialize them and we might fail to commercialize products or programs for which a suitable collaborator cannot be found. If we fail to achieve successful collaborations, our operating results and financial condition will be materially and adversely affected.
We depend on third-party contractors for a substantial portion of our drug development activities and may not be able to control their work as effectively as if we performed these functions ourselves.
We outsource, and intend to continue to outsource, substantial portions of our drug development activities to third-party service providers, including manufacturing and the conduct of our clinical trials and various preclinical studies. Our agreements with third-party service providers and CROs are and will be on a study-by-study basis and typically short-term. In all cases, we expect to be able to terminate the agreements with notice and be responsible for the supplier’s previously incurred costs.
Because we rely on third parties, our internal capacity to perform these functions is limited. Outsourcing these functions involves risk that third parties may not perform to our standards, may not produce results in a timely manner or may fail to perform at all. Even if we outsource activities, in most cases regulators will hold us responsible for the compliance of the activities performed, and hold us responsible for oversight and monitoring of the activities. In addition, the use of third-party service providers requires us to disclose our proprietary information to these parties, which could increase the risk that this information will be misappropriated. There are a limited number of third-party service providers that have the expertise required to achieve our business objectives. Identifying, qualifying and managing performance of third-party service providers can be difficult and time consuming and could cause delays in our development programs. We currently have a small number of employees devoted to clinical development activities, which limits the internal resources we have available to identify and monitor our third-party providers. To the extent we are unable to identify, retain and successfully manage the performance of third-party service providers in the future, our business may be adversely affected.
We have no experience manufacturing product candidates on a large clinical or commercial scale. As a result, we are and will be dependent on third parties for the manufacture of SCY-078 and any future product candidates we may seek to develop, and if we experience problems with any of these third parties, the commercial manufacturing of SCY-078 and any future product candidates we may seek to develop could be delayed.
If SCY-078 is approved, the inability to manufacture sufficient commercial supplies of the drug product could adversely affect product commercialization. We do not currently have any agreements with third-party manufacturers for the long-term commercial supply of our product candidates, including SCY-078. We may encounter technical difficulties or delays in the transfer of SCY-078 manufacturing on a commercial scale to a third-party manufacturer, or may be unable to enter into agreements for commercial supply with third-party manufacturers, or may be unable to do so on acceptable terms.
We may not be able to establish additional sources of supply for SCY-078 and any future product candidates we may seek to develop. These suppliers are subject to regulatory requirements covering manufacturing, testing, quality control and record keeping relating to product candidates and are also subject to ongoing inspections by the regulatory agencies. Failure by any of our suppliers to comply with applicable regulations may result in long delays and interruptions to our product candidate supply while we seek to secure another supplier that meets all regulatory requirements.
Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured product candidates ourselves, including:
| |
• | the possible breach of the manufacturing agreements or violation of regulatory standards by the third parties because of factors beyond our control; and |
| |
• | the possibility of termination or nonrenewal of the agreements by the third parties because of our breach of the manufacturing agreement or based on their own business priorities. |
Any of these factors could result in delays or higher costs in connection with our clinical trials, regulatory submissions, required approvals or commercialization of SCY-078 and any future product candidates we may seek to develop.
If we fail to establish or lose our relationships with CROs, our drug development efforts could be delayed.
We are substantially dependent on third-party vendors and CROs for preclinical studies and clinical trials related to our drug discovery and development efforts. If we fail to establish or lose our relationship with any one or more of these providers, we could experience a significant delay in both identifying another comparable provider and then contracting for its services, which could adversely affect our development efforts. We may be unable to retain an alternative provider on reasonable terms,
or at all. Even if we locate an alternative provider, it is likely that this provider will need additional time to respond to our needs and may not provide the same type or level of services as the original provider. In addition, any contract research organization that we retain will be subject to the FDA’s regulatory requirements and similar foreign standards and we do not have control over compliance with these regulations by these providers. Consequently, if these practices and standards are not adhered to by these providers, the development and commercialization of SCY-078 and any future product candidates we may seek to develop could be delayed, which could severely harm our business and financial condition.
Risks Relating to Our Intellectual Property
We were dependent on Merck for the establishment of our intellectual property rights related to SCY-078, and if Merck did not establish our intellectual property rights with sufficient scope to protect SCY-078, we may have limited or no ability to assert intellectual property rights to SCY-078.
Under our agreement with Merck, Merck was responsible for establishing the intellectual property rights to SCY-078. As we were not responsible for the establishment of our intellectual property rights to SCY-078, we have less visibility into the strength of our intellectual property rights to SCY-078 than if we had been responsible for the establishment of these rights. If Merck did not establish those rights such that they are of sufficient scope to protect SCY-078, then we may not be able to prevent others from using or commercializing SCY-078, and others may be able to assert intellectual property rights in SCY-078 and prevent us from further pursuing the development and commercialization of SCY-078.
It is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection.
Our commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of SCY-078 and any future product candidates we may seek to develop and the methods used to manufacture them, as well as successfully defending these patents against third-party challenges. Our ability to stop third parties from making, using, selling, offering to sell or importing SCY-078 and any future product candidates we may seek to develop is dependent upon the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities.
The patent positions of pharmaceutical companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No absolute policy regarding the breadth of claims allowed in pharmaceutical patents has emerged to date in the United States or in many foreign jurisdictions. Changes in either the patent laws or in interpretations of patent laws in the United States and foreign jurisdictions may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be enforced in the patents that we currently own or that may be issued from the applications we have filed or may file in the future or that we have licensed or may license from third parties, including Merck for SCY-078. Further, if any patents we obtain or license are deemed invalid or unenforceable, it could impact our ability to commercialize or license our technology.
The degree of future protection for our proprietary rights is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| |
• | others may be able to make compounds that are similar to SCY-078 and any future product candidates we may seek to develop but that are not covered by the claims of our patents; |
| |
• | if we encounter delays in our clinical trials, the period of time during which we could market our drug candidates under patent protection would be reduced; |
| |
• | we might not have been the first to conceive, make or disclose the inventions covered by our patents or pending patent applications; |
| |
• | we might not have been the first to file patent applications for these inventions; |
| |
• | any patents that we obtain may be invalid or unenforceable or otherwise may not provide us with any competitive advantages; or |
| |
• | the patents of others may have a material adverse effect on our business. |
Due to the patent laws of a country, or the decisions of a patent examiner in a country, or our own filing strategies, we may not obtain patent coverage for all of the product candidates that may be disclosed or methods involving these candidates that may be disclosed in the parent patent application. We plan to pursue divisional patent applications and/or continuation patent applications in the United States and many other countries to obtain claim coverage for inventions that were disclosed but not claimed in the parent patent application, but may not succeed in these efforts.
Composition of matter patents on the active pharmaceutical ingredient are generally considered to be the strongest form of intellectual property protection for pharmaceutical products, as such patents generally provide protection without regard to
any method of use. We cannot be certain that the claims in our patent applications covering composition-of-matter of our drug candidates will be considered patentable by the U.S. Patent and Trademark Office, or USPTO, courts in the United States or by the patent offices and courts in foreign countries. Method of use patents protect the use of a product for the method recited in the claims. This type of patent does not prevent a competitor from making and marketing a product that is identical to our product for an indication that is outside the scope of the patented method. Moreover, even if competitors do not actively promote their product for our targeted indications, physicians may prescribe these products “off-label.” Although off-label prescriptions may infringe or contribute to or induce the infringement of method of use patents, the practice is common and such infringement is difficult to prevent or prosecute. Interference or derivation proceedings provoked by third parties or brought by the USPTO may be necessary to determine the priority of inventions with respect to our patents or patent applications or those of our collaborators or licensors. An unfavorable outcome could require us to cease using the related technology or to attempt to license rights to it from the prevailing party. Our business could be harmed if the prevailing party does not offer us a license on commercially reasonable terms. Litigation, interference, or derivation proceedings may fail, resulting in harm to our business, and, even if successful, may result in substantial costs and distract our management and other employees.
There have been numerous changes to the patent laws and proposed changes to the rules of the USPTO, which may have a significant impact on our ability to protect our technology and enforce our intellectual property rights. For example, in September 2011, President Obama signed the America Invents Act that codifies several significant changes to the U.S. patent laws, including, among other things, changing from a “first to invent” to a “first inventor to file” system, limiting where a patent holder may file a patent suit, replacing interference or “first to invent” proceedings with derivation proceedings and creating inter partes review and post-grant opposition proceedings to challenge the validity of patents after they have been issued. The effects of these changes are currently unclear as the USPTO only recently has adopted regulations implementing the changes, the courts have yet to address most of these provisions, and the applicability of the act and new regulations on specific patents and patent applications discussed herein have not been determined and would need to be reviewed.
Periodic maintenance fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can in many cases be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction. Noncompliance events that could result in abandonment or lapse of a patent or patent application include, but are not limited to, failure to respond to official actions within prescribed time limits, non-payment of fees and failure to properly legalize and submit formal documents. In such an event, our competitors might be able to enter the market in the relevant country or region, which could have a material adverse effect on our business.
We also rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, licensees, licensors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information such that our competitors may obtain it. Enforcing a claim that a third party illegally obtained and is using any of our trade secrets is expensive and time consuming, and the outcome is unpredictable. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how, such as new therapies, including therapies for the indications we are targeting. If others seek to develop similar therapies, their research and development efforts may inhibit our ability to conduct research in certain areas and to expand our intellectual property portfolio, and also have a material adverse effect on our business.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and we may be unable to enforce or protect our rights to, or use, our technology.
If we choose to go to court to stop another party from using the inventions claimed in any patents we obtain, that individual or company has the right to ask the court to rule that such patents are invalid or should not be enforced. These lawsuits are expensive and would consume time and resources and divert the attention of managerial and scientific personnel even if we were successful in stopping the infringement of such patents or sustaining their validity and enforceability. In addition, there is a risk that the court will decide that such patents are not valid or that we do not have the right to enforce them. There is also the risk that, even if the validity of such patents is upheld, the court will refuse to stop the other party on the grounds that such other party’s activities do not infringe such patents. In addition, the United States Court of Appeals for the Federal Circuit and the Supreme Court of the United States continue to address issues under the United States patent laws, and the decisions of those and other courts could adversely affect our ability to sustain the validity of our issued or licensed patents and obtain new patents.
Furthermore, a third party may claim that we or our manufacturing or commercialization partners or customers are using inventions covered by the third party’s patent rights and may go to court to stop us or our partners and/or customers from engaging in our operations and activities, including making or selling SCY-078 and any future product candidates we may seek to develop. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and scientific personnel. There is a risk that a court would decide that we or our commercialization partners or customers are infringing the third party’s patents and would order us or our partners or customers to stop the activities covered by the patents. In that event, we or our commercialization partners or customers may not have a viable way around the patent and may need to halt commercialization or use of the relevant product. In addition, there is a risk that a court will order us or our partners or customers to pay the other party damages for having violated the other party’s patents or obtain one or more licenses from third parties, which may be impossible or require substantial time and expense. We cannot predict whether any license would be available at all or whether it would be available on commercially reasonable terms. Furthermore, even in the absence of litigation, we may need to obtain licenses from third parties to advance our research or allow commercialization of our drug candidates, and we have done so from time to time. We may fail to obtain any of these licenses at a reasonable cost or on reasonable terms, if at all. In such events, we would be unable to further develop and commercialize one or more of our drug candidates, which could harm our business significantly. In the future, we may agree to indemnify our commercial partners and/or customers against certain intellectual property infringement claims brought by third parties which could increase our financial expense, increase our involvement in litigation and/or otherwise materially adversely affect our business.
Because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation, which could adversely affect our intellectual property rights and our business. In addition, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock.
The pharmaceutical and biotechnology industries have produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. For example, we are aware of the existence of other patents relating to the treatment of Hepatitis C Virus which, if the compositions or methods claimed in the patents we assigned to Waterstone are practiced and determined to infringe, may limit Waterstone’s ability to fully commercialize SCY-635 and, as a result, may limit potential milestone and royalty payments due to us from Waterstone upon commercialization of SCY-635. If we are sued for patent infringement, we would need to demonstrate that our products or methods either do not infringe the patent claims of the relevant patent or that the patent claims are invalid or unenforceable, and we may not be able to do this. Proving invalidity or unenforceability is difficult. For example, in the United States, proving invalidity requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
Because some patent applications in the United States may be maintained in secrecy until the patents are issued, because patent applications in the United States and many foreign jurisdictions are typically not published until eighteen months after filing, because searches and examinations of patent applications by the USPTO and other patent offices may not be comprehensive, and because publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our patents or pending applications. Our competitors may have filed, and may in the future file, patent applications and may have obtained patents covering technology similar to ours. Any such patents or patent application may have priority over our patent applications, which could further require us to obtain or license rights to issued patents covering such technologies. If another party has obtained a U.S. patent or filed a U.S. patent application on inventions similar to ours, we may have to participate in a proceeding before the USPTO or in the courts to determine which patent or application has priority. The costs of these proceedings could be substantial, and it is possible that our application or patent could be determined not to have priority, which could adversely affect our intellectual property rights and business.
We have received confidential and proprietary information from collaborators, prospective licensees and other third parties. In addition, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies. We may be subject to claims that we or our employees, consultants or independent contractors have improperly used or disclosed confidential information of these third parties or our employees’ former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial cost and be a distraction to our management and employees. If we are not successful, our ability to continue our operations and our business could be materially, adversely affected.
Some of our competitors may be able to sustain the costs of complex intellectual property litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and
continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations, on our ability to hire or retain employees, or otherwise on our business.
Risks Related to Employee Matters and Managing Growth
We may not be able to manage our business effectively if we are unable to attract and retain key personnel.
We may not be able to attract or retain qualified management, finance, scientific and clinical personnel in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses, due in part to the relocation of our corporate and operational headquarters to Jersey City, New Jersey. Stock-based awards are critical to our ability to recruit, retain and motivate highly skilled talent. However, the trading price of our common stock as listed on the NASDAQ Global Market has traded at or below the exercise price of a significant portion of the stock options currently held by our executive officers and key employees. This may reduce the retention value of these options and we may need to grant additional stock options, make further amendments to the terms of existing option awards, or provide alternative compensation and retention programs to continue to retain our employees, especially our key employees and executive officers. If we are not able to attract and retain necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy. During 2015 our management team changed significantly, including new persons in the roles of President and Chief Executive Officer, Chief Financial Officer, and Chief Medical Officer. As a result, we have had significant management turnover, and if we are unable to retain our current executive officers and key employees our ability to implement our business strategy successfully could be seriously harmed.
We may need to expand our operations and increase the size of our company, and we may experience difficulties in managing growth.
As we advance SCY-078 through preclinical studies, clinical trials and commercialization, we will need to increase our product development, scientific, marketing, sales and administrative headcount to manage these efforts. Our management, personnel and systems currently in place may not be adequate to support this future growth. Our need to effectively manage our operations, growth and various projects requires that we:
| |
• | successfully attract and recruit new employees with the expertise and experience we will require; |
| |
• | manage our clinical programs effectively, which we anticipate being conducted at numerous clinical sites; |
| |
• | develop a marketing and sales infrastructure; and |
| |
• | continue to develop our operational, financial and management controls, reporting systems and procedures. |
If we are unable to successfully manage this growth, our business may be adversely affected.
Other Risks Relating to Our Business
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for a product candidate and may have to limit its commercialization.
The use of product candidates in clinical trials and the sale of any products for which we may obtain marketing approval expose us to the risk of product liability claims. Product liability claims may be brought against us or our partners by participants enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling products. If we cannot successfully defend ourselves against any such claims, we would incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
| |
• | withdrawal of clinical trial participants; |
| |
• | termination of clinical trial sites or entire trial programs; |
| |
• | costs of related litigation; |
| |
• | substantial monetary awards to patients or other claimants; |
| |
• | decreased demand for product candidates and loss of revenue; |
| |
• | impairment of our business reputation; |
| |
• | diversion of management and scientific resources from our business operations; and |
| |
• | the inability to commercialize product candidates. |
We have obtained limited product liability insurance coverage for our clinical trials domestically and in selected foreign countries where we are conducting clinical trials. Our coverage is currently limited to $5.0 million per occurrence and $5.0 million in the aggregate per year, as well as additional local country product liability coverage for trials conducted outside of the United States as required by the local country regulations. As such, our insurance coverage may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to product liability. We intend to expand our insurance coverage for products to include the sale of commercial products if we obtain marketing approval for product candidates, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could decrease our cash available to develop SCY-078 and any future product candidates we may seek to develop and adversely affect our business.
Our internal computer systems, or those used by our contract research organizations or other contractors or consultants, may fail or suffer security breaches.
Despite the implementation of security measures, our internal computer systems and those of our contract research organizations and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. While we have not experienced any such system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our product candidate development programs. For example, the loss of clinical study data from completed or ongoing clinical studies for a product candidate could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach was to result in a loss of or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development of any product candidates could be delayed.
Our former Services Business operations involved, and the operations of our vendors may involve, the use of hazardous materials, which could subject us to significant liabilities.
Research and development processes performed by our former Services Business, and research and development processes that we contract with vendors to perform, involve the controlled use of hazardous materials, including chemicals. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these materials. Individuals exposed to these hazardous materials could attempt to assert civil liability against us either as the former operator of the Services Business or as the contractor for services performed by our vendors. We have general liability insurance coverage of up to $1.0 million per occurrence, with an annual aggregate limit of $2.0 million, which excludes pollution liability. This coverage may not be adequate to cover all claims. Furthermore, if we were to be held liable for a claim involving biological or hazardous materials, this liability could exceed our insurance coverage, if any, and our other financial resources.
Our insurance policies are expensive and protect us only from some business risks, which will leave us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. Some of the policies we currently maintain include general liability, employment practices liability, property, auto, workers’ compensation, products liability and directors’ and officers’ insurance. We do not know, however, if we will be able to maintain existing insurance with adequate levels of coverage. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our cash position and results of operations.
Our research and development activities could be affected or delayed as a result of possible restrictions on animal testing.
Certain laws and regulations require us to test our product candidates on animals before initiating clinical trials involving humans. Animal testing activities have been the subject of controversy and adverse publicity. Animal rights groups and other organizations and individuals have attempted to stop animal testing activities by pressing for legislation and regulation in these areas and by disrupting these activities through protests and other means. To the extent the activities of these groups are successful, our research and development activities may be interrupted, delayed or become more expensive.
Risks Relating to Owning Our Common Stock
The market price of our common stock may be highly volatile.
The trading price of our common stock may be volatile. The following factors, in addition to other factors described in this “Risk Factors” section and elsewhere in this report, may have a significant impact on the market price of our common stock:
| |
• | the results of our preclinical testing or clinical trials; |
| |
• | the ability to obtain additional funding; |
| |
• | any delay in filing an NDA or similar foreign applications for SCY-078 and any future product candidate we may seek to develop or any adverse development or perceived adverse development with respect to the FDA’s review of that NDA or a foreign regulator’s review of a similar applications; |
| |
• | maintenance of our existing collaborations or ability to enter into new collaborations; |
| |
• | our collaboration partners’ election to develop or commercialize product candidates under our collaboration agreements or the termination of any programs under our collaboration agreements; |
| |
• | any intellectual property infringement actions in which we or our licensors and collaboration partners may become involved; |
| |
• | our ability to successfully develop and commercialize future product candidates; |
| |
• | changes in laws or regulations applicable to future products; |
| |
• | adverse regulatory decisions; |
| |
• | introduction of new products, services or technologies by our competitors; |
| |
• | achievement of financial projections we may provide to the public; |
| |
• | achievement of the estimates and projections of the investment community; |
| |
• | the perception of the pharmaceutical industry by the public, legislatures, regulators and the investment community; |
| |
• | announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us, our collaboration partners or our competitors; |
| |
• | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
| |
• | legislation or regulation that mandates or encourages the use of generic products; |
| |
• | additions or departures of key scientific or management personnel; |
| |
• | significant lawsuits, including patent or stockholder litigation; |
| |
• | changes in the market valuations of similar companies; |
| |
• | general economic and market conditions and overall fluctuations in the U.S. equity markets; |
| |
• | sales of our common stock by us, our executive officers and directors or our stockholders in the future; and |
| |
• | trading volume of our common stock. |
In addition, companies trading in the stock market in general, and the NASDAQ Global Market in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance.
Our executive officers, directors and principal stockholders own a significant percentage of our stock and will be able to exert significant control over matters submitted to our stockholders for approval.
As of February 29, 2016, our executive officers, directors and stockholders who own more than 5% of our outstanding common stock, together own shares representing a substantial portion of our outstanding common stock. Therefore, these stockholders will have the ability to influence us through this ownership position. These stockholders may be able to influence matters requiring stockholder approval. For example, these stockholders, acting together, may be able to control elections of directors, amendments to our organizational documents, or approval of any merger, sale of assets, or other major corporate action. This may prevent or discourage unsolicited acquisition proposals or offers for our common stock that you may believe are in your best interest as one of our stockholders.
We may identify material weaknesses in our internal controls over financial reporting.
Maintaining effective internal controls over financial reporting is necessary for us to produce accurate financial statements on a timely basis. We have previously identified material weaknesses in our internal control over financial reporting and, although all such material weaknesses were remediated as of December 31, 2014, we may again identify material weaknesses in the future. Management continues to devote significant time, attention, and resources to maintaining and improving our internal controls. We expect to continue to incur costs associated with implementing appropriate processes and internal controls, which could include new employee compensation costs and fees for additional audit and consulting services, which could negatively affect our financial condition and operating results.
The requirements associated with being a public company will require significant company resources and management attention.
We completed our IPO in May 2014 and have become subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, or the Exchange Act, the Sarbanes-Oxley Act of 2002, or the Sarbanes-Oxley Act, the listing requirements of the NASDAQ Global Market and other applicable securities rules and regulations. The Exchange Act requires that we file annual, quarterly and current reports with respect to our business and financial condition and maintain effective disclosure controls and procedures and internal control over financial reporting. In addition, subsequent rules implemented by the SEC and the NASDAQ Stock Market may also impose various additional requirements on public companies. As a result, we will incur additional legal, accounting and other expenses that we did not incur as a nonpublic company, particularly after we are no longer an “emerging growth company” as defined in the JOBS Act. Further, the need to establish the corporate infrastructure demanded of a public company may divert management’s attention from implementing our growth strategy. We have made, and will continue to make, changes to our corporate governance standards, disclosure controls and financial reporting and accounting systems to meet our reporting obligations. However, the measures we take may not be sufficient to satisfy our obligations as a public company, which could subject us to delisting of our common stock, fines, sanctions and other regulatory action and potentially civil litigation.
Section 404(a) of the Sarbanes-Oxley Act requires annual management assessments of the effectiveness of our internal control over financial reporting, starting with the second annual report that we would expect to file with the SEC, and we are required to disclose material changes made in our internal controls and procedures on a quarterly basis. Company responsibilities required by the Sarbanes-Oxley Act include establishing corporate oversight and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent financial fraud. However, our independent registered public accounting firm will not be required to attest to the effectiveness of our internal control over financial reporting pursuant to Section 404(b) of the Sarbanes-Oxley Act until the later of the year following our first annual report required to be filed with the SEC or the date we are no longer an “emerging growth company” as defined in the JOBS Act, because we are taking advantage of the exemptions contained in the JOBS Act.
If we identify one or more material weaknesses in our internal control over financial reporting, we will be unable to assert that our internal control over financial reporting is effective. We cannot assure you that there will not be material weaknesses in our internal control over financial reporting in the future. Any failure to maintain internal control over financial reporting could severely inhibit our ability to accurately report our financial condition, results of operations or cash flows. If we are unable to achieve effective internal control over financial reporting, or if our independent registered public accounting firm determines we have a material weakness in our internal control over financial reporting, we could lose investor confidence in the accuracy and completeness of our financial reports, the market price of our common stock could decline, and we could be subject to sanctions or investigations by the NASDAQ Stock Market, the SEC or other regulatory authorities. Failure to remedy
any material weakness in our internal control over financial reporting, or to implement or maintain other effective control systems required of public companies, could also restrict our future access to the capital markets.
The recently enacted JOBS Act will allow us to postpone the date by which we must comply with some of the laws and regulations intended to protect investors and to reduce the amount of information we provide in our reports filed with the SEC, which could undermine investor confidence in our company and adversely affect the market price of our common stock.
For so long as we remain an “emerging growth company” as defined in the JOBS Act, we may take advantage of certain exemptions from various requirements that are applicable to public companies that are not “emerging growth companies” including:
| |
• | the provisions of Section 404(b) of the Sarbanes-Oxley Act requiring that our independent registered public accounting firm provide an attestation report on the effectiveness of our internal control over financial reporting; |
| |
• | the “say on pay” provisions, requiring a non-binding stockholder vote to approve compensation of certain executive officers, and the “say on golden parachute” provisions, requiring a non-binding stockholder vote to approve golden parachute arrangements for certain executive officers in connection with mergers and certain other business combinations, of the Dodd-Frank Act and some of the disclosure requirements of the Dodd-Frank Act relating to compensation of our chief executive officer; |
| |
• | the requirement to provide detailed compensation discussion and analysis in proxy statements and reports filed under the Exchange Act, and instead provide a reduced level of disclosure concerning executive compensation; and |
| |
• | any rules that may be adopted by the Public Company Accounting Oversight Board requiring mandatory audit firm rotation or a supplement to the auditor’s report on the financial statements. |
We currently intend to take advantage of some of the reduced regulatory and reporting requirements that will be available to us under the JOBS Act so long as we qualify as an “emerging growth company.”
Future sales and issuances of our common stock or rights to purchase common stock could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations. To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. These sales may also result in new investors gaining rights superior to our existing stockholders.
Because we do not intend to declare cash dividends on our shares of common stock in the foreseeable future, stockholders must rely on appreciation of the value of our common stock for any return on their investment.
We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends in the future. As a result, we expect that only appreciation of the price of our common stock, if any, will provide a return to our investors for the foreseeable future. Investors seeking cash dividends should not invest in our common stock.
If securities or industry analysts do not publish or cease publishing research or reports about us, our business or our market, or if they change their recommendations regarding our common stock adversely, the price of our common stock and trading volume could decline.
The trading market for our common stock will be influenced by the research and reports that securities or industry analysts may publish about us, our business, our market or our competitors. If any of the analysts who may cover us change their recommendation regarding our common stock adversely, or provide more favorable relative recommendations about our competitors, the price of our common stock would likely decline. If any analyst who may cover us were to cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which in turn could cause the price of our common stock or trading volume to decline.
We may be subject to securities litigation, which is expensive and could divert management attention.
Our share price may be volatile, and in the past, companies that have experienced volatility in the market price of their stock have been subject to securities class action litigation. We may be the target of this type of litigation in the future.
Litigation of this type could result in substantial costs and diversion of management’s attention and resources, which could seriously harm our business. Any adverse determination in litigation could also subject us to significant liabilities.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and our bylaws may delay or prevent an acquisition of us, including the ability of our board of directors to establish new series of preferred stock and issue shares of these new series, which could be used by our board of directors to oppose a hostile takeover attempt, which some stockholders may believe would be in the best interests of stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management, including the elimination of cumulative voting, inability of our stockholders to call special meetings or take action by written consent, ability of our board of directors to fill board vacancies, and ability of our board of directors to determine the size of the board of directors. In addition, we are subject to Section 203 of the Delaware General Corporation Law, which generally prohibits stockholders owning in excess of 15% of our outstanding voting stock from merging or combining with us. Finally, our charter documents establish advance notice requirements for nominations for election to our board of directors and for proposing matters that can be acted upon at stockholder meetings. Although we believe these provisions together provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if the offer may be considered beneficial by some stockholders.
| |
ITEM 1B. | UNRESOLVED STAFF COMMENTS |
None.
We sublease 10,141 square feet of space located at 101 Hudson Street, Suite 3610, Jersey City, New Jersey, which consists solely of office space. The term of the sublease is scheduled to expire on July 30, 2018.
| |
ITEM 3. | LEGAL PROCEEDINGS |
None.
| |
ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
PART II
| |
ITEM 5. | MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES |
Market Information
Our common stock began trading on the NASDAQ Global Market on May 2, 2014 under the symbol “SCYX.” Prior to such time, there was no public market for our common stock. The following table sets forth the high and low sales prices per share of our common stock as reported on the NASDAQ Global Market for the periods indicated. Such quotations represent inter-dealer prices without retail markup, markdown or commission and may not necessarily represent actual transactions.
|
| | | | | | | | |
Year Ended December 31, 2014 | | High | | Low |
Second Quarter | | $ | 9.89 |
| | $ | 7.78 |
|
Third Quarter | | $ | 8.34 |
| | $ | 5.10 |
|
Fourth Quarter | | $ | 11.98 |
| | $ | 5.70 |
|
|
| | | | | | | | |
Year Ended December 31, 2015 | | High | | Low |
First Quarter | | $ | 15.00 |
| | $ | 7.09 |
|
Second Quarter | | $ | 10.05 |
| | $ | 7.50 |
|
Third Quarter | | $ | 9.10 |
| | $ | 6.29 |
|
Fourth Quarter | | $ | 7.69 |
| | $ | 5.51 |
|
Stockholders
As of March 1, 2016, there were approximately 118 stockholders of record of our common stock, which excludes stockholders whose shares were held in nominee or street name by brokers. The actual number of common stockholders is greater than the number of record holders, and includes stockholders who are beneficial owners, but whose shares are held in street name by brokers and other nominees. This number of holders of record also does not include stockholders whose shares may be held in trust by other entities.
Dividend Policy
We have never declared or paid any cash dividends on our common stock. We currently intend to retain all available funds and any future earnings to support our operations and finance the growth and development of our business. We do not intend to pay cash dividends on our common stock for the foreseeable future. Any future determination related to our dividend policy will be made at the discretion of our board of directors and will depend upon, among other factors, our results of operations, financial condition, capital requirements, contractual restrictions, business prospects and other factors our board of directors may deem relevant.
Use of Proceeds
On May 2, 2014, our registration statement on Form S-1 (File No. 333-194192) was declared effective for our initial public offering of 6,200,000 shares of our common stock at a price of $10.00 per share for aggregate gross proceeds of $62.0 million to us. As a result of our IPO, which closed on May 7, 2014, we received net proceeds of approximately $54.6 million after deducting underwriting discounts and commissions of $3.3 million and offering expenses payable by us of $4.1 million.
There has been no material change in the planned use of proceeds from our initial public offering as described in our prospectus effective May 2, 2014, filed with the SEC pursuant to Rule 424(b) of the Securities Act. Through December 31, 2015, $45.6 million of the net proceeds had been used for the purposes set forth in our prospectus, including $15.0 million to pay off the balance and all accrued interest on our credit facility with HSBC Bank on May 7, 2014, and $30.6 million for the development of our lead product candidate SCY-078 and to fund working capital, capital expenditures and other general corporate purposes.
Recent Sales of Unregistered Securities
None.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
We did not purchase any of our registered securities during the fourth quarter of 2015.
| |
ITEM 6. | SELECTED FINANCIAL DATA |
As a Smaller Reporting Company until December 31, 2015, we are not required to provide the disclosure required by this Item until our next Annual Report on Form 10-K.
| |
ITEM 7. | MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
Operating results for the year ended December 31, 2015, are not necessarily indicative of results that may occur in future interim periods or future fiscal years. Some of the statements in this “Management’s Discussion and Analysis of Financial Condition and Results of Operations” are forward-looking statements. These forward-looking statements are based on management’s beliefs and assumptions and on information currently available to our management and involve significant elements of subjective judgment and analysis. Words such as “expects,” “will,” “anticipates,” “targets,” “goals,” “projects,” “intends,” “plans,” “believes,” “seeks,” “estimates,” “potential,” “should,” “could,” variations of such words, and similar expressions are intended to identify forward-looking statements. Our actual results and the timing of events may differ significantly from the results discussed in the forward-looking statements. Factors that might cause such a difference include those discussed under the caption “Special Note Regarding Forward Looking Statements” and in “Risk Factors” and elsewhere in this Annual Report on Form 10-K. These and many other factors could affect our future financial and operating results. We undertake no obligation to update any forward-looking statement to reflect events after the date of this Annual Report.
Overview
SCYNEXIS is a pharmaceutical company committed to the development and commercialization of novel anti-infectives to address significant unmet therapeutic needs. We are developing our lead product candidate, SCY-078, as a novel oral and intravenous (IV) drug for the treatment of several fungal infections, including serious and life-threatening invasive fungal infections. SCY-078 is a novel and structurally distinct glucan synthase inhibitor that has been shown to be effective in vitro and in vivo in animal studies against a broad range of Candida and Aspergillus species, including drug-resistant strains, and we are continuing to conduct additional in vitro and in vivo studies to further characterize the spectrum of activity of SCY-078. Candida and Aspergillus species are the fungi responsible for approximately 85% of all invasive fungal infections in the United States and Europe. We have completed multiple Phase 1 studies with the oral formulation of SCY-078 and we are currently conducting our first Phase 1 study with the IV formulation of SCY-078. We are also conducting two Phase 2 studies with the oral formulation of SCY-078:
| |
• | the first study is evaluating the safety, tolerability, and pharmacokinetics of SCY-078 as oral step-down treatment in patients initially treated with IV echinocandin therapy for invasive Candida infections and; |
| |
• | the second study is evaluating the safety and efficacy of orally administered SCY-078 for the treatment of vulvovaginal candidiasis (VVC). |
SCY-078 holds both Fast Track and Qualified Infections Disease Product (QIDP) designations for the IV and oral formulations for the indications of invasive candidiasis (including candidemia) and invasive aspergillosis. We expect to complete and report top line data associated with our two Phase 2 studies, our Phase 1 study and our additional in vitro and in vivo studies by the end of the second quarter of 2016.
As a spinout from Aventis S.A., or Aventis in 2000, we began as a chemistry and animal health services company, providing contract research services to third parties. Through the provision of these contract research and development services, we built significant expertise in parasitic infections and drug discovery, including expanded animal health capabilities. This contract research and development services business, which we refer to as our “Services Business,” generated substantially all of our revenue until we completed the sale of the Services Business to Accuratus Lab Services, Inc. in July 2015, as described further in “Recent Developments” below. Since our formation, in addition to SCY-078 and related antifungal compounds, we have discovered a number of proprietary compounds, including those within our cyclophilin inhibitor platform. We are currently focusing our resources on the development of SCY-078. In the future, we may develop other assets within our proprietary portfolio of antifungal or cyclophilin inhibitor compounds either in-house or through collaborations with strategic development partners. Additionally, we may assess external opportunities to expand our clinical pipeline.
We have operated as a public entity since we completed our initial public offering in May 2014, which we refer to as our IPO. We also completed a follow-on public offering of our common stock in April 2015. As of December 31, 2015, we had received an aggregate of $92.6 million in net proceeds from the issuance of our common stock in these two offerings. Our principal source of liquidity is cash and cash equivalents, which totaled $47.0 million as of December 31, 2015.
We have incurred net losses since our inception, including the year ended December 31, 2015. As of December 31, 2015, our accumulated deficit was $150.1 million. We anticipate that we will continue to incur losses for at least the next several years. We expect that our research and development expenses will continue to increase as we continue to execute our research and drug development strategy. We also expect that we will continue to incur selling, general and administrative expenses to support our public reporting company operations. As a result, we will need additional capital to fund our operations, which we may obtain through one or more of equity offerings, debt financings, or other non-dilutive third-party funding (e.g., grants), strategic alliances and licensing or collaboration arrangements. We may offer shares of our common stock pursuant to our Form S-3 shelf registration statement filed with the SEC on October 30, 2015 and declared effective on November 16, 2015, including the related at-the-market facility filed under the Sales Agreement Prospectus on the same date.
We are an emerging growth company. Under the Jumpstart Our Business Startups Act of 2012, or JOBS Act, emerging growth companies can delay adopting new or revised accounting standards until such time that those standards apply to private companies. We have irrevocably elected not to adopt this exemption from new or revised accounting standards, and therefore, we will be subject to the same new or revised accounting standards as other public companies that are not “emerging growth companies.”
Recent Developments
SCY-078 Development
We are conducting a multicenter Phase 2 study with primary endpoints of safety, tolerability, and pharmacokinetics of the oral formulation of SCY-078 as step-down treatment in patients initially treated with echinocandin therapy for invasive Candida infections. We have opened new investigational sites in the U.S. and in Latin America and we are in the process of opening more sites in these regions and in Europe. Based on the data collected on the enrolled patients, together with the data from our recently completed Phase 1 biocomparison study, we expect to achieve the primary objectives of the study with fewer patients than originally planned and to report top line data by the end of the second quarter of 2016.
We are conducting a multicenter Phase 2 study with primary endpoints of safety and efficacy of the oral formulation of SCY-078 in patients with VVC. We expect to complete the study and to report top line data by the end of the second quarter of 2016. We expect the data from this study to provide a confirmation of the potential therapeutic effect of orally administered SCY-078 in a clinical condition caused by Candida spp. and, along with the other clinical and nonclinical data from ongoing and planned activities, will contribute to the package of information that will support subsequent phases of development.
We are conducting a single-rising-dose Phase 1 study to evaluate the safety, tolerability and pharmacokinetics of SCY-078 administered as an intravenous infusion in healthy subjects. We expect to complete the study and to report results by the end of the second quarter of 2016.
We recently completed a Phase 1 biocomparison study of a new, well tolerated citrate salt formulation of SCY-078 that has a comparable pharmacokinetic profile and potential formulation advantages over the previously used phosphate salt formulation. Further development activities for both the oral and IV formulations of SCY-078 are planned with the citrate salt.
Both the oral and IV formulations of SCY-078 have been granted QIDP designation and fast track designation by the FDA for both invasive candidiasis and invasive aspergillosis. The fast track designation, coupled with the QIDP designation, allows for a potentially accelerated path to approval and underscores the FDA's understanding of the critical need for new and varied treatments for life-threatening invasive fungal infections.
April 2015 Follow-On Public Offering
On April 28, 2015, we completed a follow-on public offering (the "April 2015 Offering") of our common stock. We sold an aggregate of 5,376,622 shares of common stock at a public offering price of $7.70 per share. Net proceeds to us were approximately $38.0 million, after deducting underwriting discounts and commissions and offering expenses of approximately $3.4 million.
Shelf Registration Filing
On October 30, 2015, we filed a shelf registration statement on Form S-3 with the SEC, which was declared effective on November 16, 2015. The registration statement contained two prospectuses:
| |
• | a base prospectus which covers the offering, issuance and sale by us of up to a maximum aggregate offering price of $150 million of our common stock, preferred stock, debt securities and warrants, including common stock or preferred stock issuable upon conversion of debt securities, common stock issuable upon conversion of preferred stock, or common stock, preferred stock or debt securities issuable upon the exercise of warrants |
| |
• | a prospectus covering the offering, issuance and sale by us of up to a maximum aggregate offering price of $40 million of our common stock that may be issued and sold under a sales agreement with Cowen and Company, LLC (the "Sales Agreement Prospectus"). |
The common stock that may be offered, issued and sold by us under the Sales Agreement Prospectus is included in the $150 million of securities that may be offered, issued and sold by us under the base prospectus. Upon termination of the sales agreement with Cowen and Company, LLC, any portion of the $40 million included in the Sales Agreement Prospectus that is not sold pursuant to the sales agreement will be available for sale in other offerings pursuant to the base prospectus and a corresponding prospectus supplement, and if no shares are sold under the sales agreement, the full $150 million of securities may be sold in other offerings pursuant to the base prospectus.
Sale of Our Services Business
As part of our strategic objective to focus our resources on the development of SCY-078, our board of directors directed our management to explore the divestiture of our contract research and development services business (the “Services Business”), which was no longer strategic to our business and did not provide any meaningful results of operations or operating capital to fund our core strategic objective in 2015. We engaged a third party firm which assisted us in evaluating several divestiture options (i.e., a third-party sale, spin-off, management buy-out transaction, or shut-down process). On May 4, 2015, our board of directors completed its evaluation of the various divestiture options and directed management to pursue a plan to sell the Services Business to Accuratus Lab Services, Inc. ("Accuratus"), a private-equity backed process chemistry, formulation, manufacturing and analytical development services provider. In connection with this action, we met the relevant criteria for reporting the Services Business as held for sale and in discontinued operations beginning in the second quarter of 2015.
On July 21, 2015, we completed the sale of the Services Business to Accuratus pursuant to an Asset Purchase Agreement with an effective date of July 17, 2015, for an aggregate purchase price of $3.9 million, subject to a working capital adjustment of $0.8 million, which reduced the proceeds at closing. In addition, a portion of the consideration payable at closing equal to $0.5 million was withheld and is subject to an escrow for a period of 12 months from the date of closing to satisfy our indemnification obligations in connection with breaches of any representation and warranties and other customary obligations under the terms of the Asset Purchase Agreement. We have not identified any breaches or other events that would cause a reduction in the escrow funds we expect to receive. The escrow funds were recorded as a receivable included in prepaid expenses and other current assets in the accompanying balance sheets. The net cash consideration received by us upon closing in July 2015 was $2.5 million, after adjusting for the items described above and a nominal escrow fee.
As a condition to the execution of the Asset Purchase Agreement, Accuratus assumed our post-closing obligation under our prior facility lease in Durham, North Carolina and we were released from any post-obligation arising under the lease.
In connection with the adoption of the Services Business Plan described in Note 15 of the accompanying financial statements in this Form 10-K, we terminated certain employees in June 2015 (the "June 2015 Terminated Employees") who became eligible for severance benefits totaling approximately $1.0 million. We incurred these severance benefit obligations in the quarterly period ended June 30, 2015 and therefore, we recognized the expense in the quarter ended June 30, 2015, in discontinued operations in the unaudited interim statements of operations. The Services Business Plan also provided for certain amendments to the terms of the outstanding stock option awards held by the June 2015 Terminated Employees, which are described in Note 10 of the accompanying financial statements in this Form 10-K.
Also in connection with the Services Business Plan, we paid cash totaling approximately $0.2 million to certain non-executive employees of the Services Business as an incentive payment upon the closing of the sale of the Services Business in July 2015. In addition, we paid cash retention compensation payments of approximately $0.8 million in January 2016 to all Service Business employees that remained eligible pursuant to the terms of the Services Business Plan. We incurred these obligations on the date of the sale of the Services Business in July 2015; therefore, we recognized the compensation expense associated with these obligations during the quarterly period ended September 30, 2015, in discontinued operations within the accompanying statements of operations in this Form 10-K.
We had a number of licensing and collaboration agreements associated with our former Services Business that were assigned to Accuratus in conjunction with the sale of the Services Business in July 2015, including (i) a contract research and screening services agreement with Merial, a wholly owned subsidiary of Sanofi, and (ii) a license, development, and commercialization agreement with Elanco Animal Health, or Elanco, the animal health division of Eli Lilly Company. Revenue recognized pursuant to the Merial and Elanco agreements during the year ended December 31, 2015 was $2.1 million and $1.7
million, respectively, and collectively accounted for approximately 52% of total revenue in discontinued operations during the year ended December 31, 2015.
The sale of our Services Business did not have a significant impact on our reported loss from continuing operations in 2015 and we do not expect it to have a significant effect on our prospective cash forecast.
Commitment to Services Agreement
On July 17, 2015, we entered into the Services Agreement with Accuratus, described in Note 18 of the accompanying audited financial statements in this Form 10-K, pursuant to which Accuratus will provide us with certain contract research and development services for 18 months (the "Initial Term") following the closing of the sale of the Services Business for a minimum purchase obligation of at least $3.3 million due from us over the Initial Term of the Services Agreement. The purpose of the Services Agreement is to replace necessary development services that were previously provided internally by our employees prior to the sale of the Services Business. The employees performing these services prior to the sale of the Services Business became employees of Accuratus in connection with the sale transaction. During the year ended December 31, 2015, we recognized $1.6 million of expense for services provided by Accuratus under the Services Agreement, which is included in research and development expense in the accompanying statements of operations in this Form 10-K.
Relocation of Headquarters and Operations, New Facilities Lease, Compensatory Arrangements with Employees
In connection with the sale of the Services Business, we relocated our corporate headquarters and operating activities to Jersey City, New Jersey. On July 13, 2015, we entered into a sublease (the "Sublease") that became effective July 22, 2015, to sublet certain premises consisting of 10,141 square feet of space (the Subleased Premises) located at 101 Hudson Street, Suite 3610, Jersey City, New Jersey from Optimer Pharmaceutical, Inc. The term of the Sublease commenced on August 1, 2015 (the Commencement Date), and is scheduled to expire on July 30, 2018. No base rent was due under the Sublease until one month after the Commencement Date. Under the Sublease, we are obligated to pay monthly base rent of approximately $25,000 per month, which amount increases by 3% annually on each anniversary of the Commencement Date. In addition, we were required to fund a security deposit with the sublandlord in the amount of $74,000.
In connection with our relocation, we designed a compensatory plan to promote the retention of services of non-executive employees supporting our continuing operations (the "Retention Plan"). The Retention Plan terms provided for certain cash compensation payments and severance payments, as well as modifications to the terms of currently outstanding stock options held by such non-executive employees. The Retention Plan provided that non-executive employees were eligible to receive cash bonuses, severance payments and related benefit premiums, provided that all employees remained employed through December 31, 2015, were not terminated for cause, and did not otherwise modify or forfeit their benefits under the Retention Plan. The Retention Plan also provided that if we and an employee agreed upon a services termination date earlier than December 31, 2015 (the "Release Date"), the employee would remain eligible for all terms of the Retention Plan. We accrued this obligation over the remaining future service period required by the employees through the earlier of the Release Date or December 31, 2015. During the year ended December 31, 2015, we recognized total expense of $1.0 million, which is included in research and development and selling, general, and administrative expenses in the accompanying statement of operations. The corresponding liability is included in accrued severance and retention obligations, current portion, in the accompanying balance sheet as of December 31, 2015. The amendments to the terms of the eligible employees' outstanding stock option awards, as provided for in the Retention Plan, are described in Note 10 of the accompanying audited annual financial statements in this Form 10-K.
Compensatory Arrangements with Former Executive Officers
Charles F. Osborne, Jr., our former chief financial officer, resigned effective June 30, 2015. Our compensation committee of the board of directors approved, and we and Mr. Osborne subsequently entered into an agreement (the "Release and Settlement Agreement") providing, a compensatory arrangement for Mr. Osborne that provided for certain payments and benefits, including (i) a cash payment of approximately $0.1 million upon his resignation on June 30, 2015; (ii) cash severance payments totaling approximately $0.2 million, which is equal to seven months of Mr. Osborne’s then base salary, paid over seven months commencing with the first payroll period following the resignation date; (iii) a payment representing a contribution Mr. Osborne can use towards continuing COBRA premiums for medical, dental, and vision group health coverage for a period up to seven months after the resignation date; and (iv) certain amendments to the terms of Mr. Osborne's outstanding stock option awards which are described in Note 10 of the accompanying audited annual financial statements in this Form 10-K. As part of this Release and Settlement Agreement, Mr. Osborne granted us a full and final release of any claims against SCYNEXIS that may have existed or arisen prior to entering into the Release and Settlement Agreement.
On July 21, 2015, Yves J. Ribeill, Ph.D., our former President and a member of our board of directors, resigned as President. Dr. Ribeill continues to serve on the board of directors. We and Dr. Ribeill entered into an agreement, effective July 21, 2015, for certain payments and benefits (the “Separation Agreement”), pursuant to which Dr. Ribeill received or will
receive: (i) a cash payment of approximately $0.1 million upon the effective date of his resignation; (ii) cash severance payments totaling approximately $0.9 million, paid over 12 months commencing with the first payroll period following the resignation date; (iii) a payment representing a contribution Dr. Ribeill can use towards continuing COBRA premiums for medical, dental, and vision group health coverage after the resignation date, and (iv) certain amendments to the terms of Dr. Ribeill's outstanding stock option awards which are described in Note 10 of the accompanying audited annual financial statements in this Form 10-K. As part of this Separation Agreement, Dr. Ribeill granted us a full and final release of any claims against SCYNEXIS that may have existed or arisen prior to the execution of the Separation Agreement.
Termination of License Agreement
In August 2012, we entered into a license agreement with Dechra Ltd. ("Dechra"), a UK listed international veterinary pharmaceutical business, granting Dechra rights to our proprietary compound, SCY-641, in the field of animal health, including dog dry eye, under which we were entitled to receive potential milestone and royalty payments. Dechra was granted worldwide animal health rights and was responsible for the remaining clinical development and commercialization of SCY-641 in the animal health field. Under the agreement, Dechra was required to use reasonable efforts to commercialize SCY-641. We received an upfront fee in 2012 and we were eligible to receive potential milestone payments as well as royalty payments on the total net sales of the product. Pursuant to the agreement, Dechra had the right to relinquish the license and terminate the agreement at any time it determined in its reasonable business judgment that it was impossible to carry out further development or marketing of the product by giving us at least six months prior written notice. In November 2015, Dechra notified us of its intention to terminate the license agreement for the development of SCY-641 effective May 2016. We do not expect the termination of this license agreement to have a significant effect on our prospective cash forecast.
Components of Operating Results
Revenue
Historically, we derived substantially all of our revenue from the provision of our contract research and development services, which were provided by our Services Business that we divested through a sale transaction in July 2015 (see “Recent Developments” above). The revenue generated from our Services Business has been presented in discontinued operations in the accompanying statements of operations and resulted in a significant decrease in our reported revenue. In addition to our contract research and development services revenue, we have received upfront and milestone payments in connection with our collaboration and licensing agreements that are associated with our continuing operations. Further, we expect that any revenue we generate will fluctuate from quarter to quarter as a result of the variability in the achievement of collaboration milestones, and the consummation of new licensing arrangements. We do not expect to generate revenue from product sales for at least the next several years. If we or our collaborators fail to complete the development of product candidates in a timely manner or obtain their regulatory approval, our ability to generate future revenue, and our results of operations and financial position, would be materially adversely affected.
Our revenue recognition policy is described within the "Critical Accounting Policies and Significant Judgments and Estimates" section below, as well as in Note 2 to our audited financial statements for the year ended December 31, 2015, included in this Form 10-K.
Research and Development Expense
Research and development expense consists of expenses incurred while performing research and development activities to discover, develop, or improve potential product candidates we seek to develop. This includes conducting preclinical studies and clinical trials, manufacturing and other development efforts, and activities related to regulatory filings for product candidates. We recognize research and development expenses as they are incurred. Our research and development expense primarily consists of:
| |
• | costs related to executing preclinical studies and clinical trials, including related drug formulation, manufacturing and other development; |
| |
• | salaries and personnel-related costs, including benefits and any stock-based compensation for personnel performing research and development functions; |
| |
• | fees paid to clinical research organizations ("CROs"), vendors, consultants and other third parties who support our product candidate development and intellectual property protection; |
| |
• | other costs in seeking regulatory approval of our products; and |
The table below summarizes the total costs incurred for each of our key research and development projects during the periods presented (dollars in thousands):
|
| | | | | | | |
| For the Years Ended December 31, |
| 2015 | | 2014 |
| |
SCY-078 | $ | 16,247 |
| | $ | 7,050 |
|
Cyclophilin Inhibitor Platform | 193 |
| | 1,237 |
|
Total Research and Development | $ | 16,440 |
| | $ | 8,287 |
|
Our SCY-078 and cyclophilin inhibitor platform projects were the only key research and development projects during the periods presented. We plan to increase our research and development expense for the foreseeable future as we continue our effort to develop SCY-078 and potentially to develop our other in-house product candidates or candidates we may acquire; subject to the availability of additional funding. We do not expect to incur any substantial research and development expenses related to our cyclophilin inhibitor platform in the near future.
The successful development of product candidates is highly uncertain. At this time, we cannot reasonably estimate the nature, timing or costs required to complete the remaining development of any product candidates. This is due to the numerous risks and uncertainties associated with the development of product candidates.
Selling, General and Administrative Expense
Selling, general and administrative expense consists primarily of salaries and personnel-related costs, including employee benefits and any stock-based compensation. This includes personnel in executive, accounting and finance, commercial, human resources and administrative support functions. Other expenses include facility-related costs not otherwise allocated to research and development expense, professional fees for accounting, auditing, tax and legal services, consulting costs for general and administrative purposes, information systems maintenance and marketing efforts.
Other (Income) Expense
Substantially all of our other (income) expense during the periods reported consists of costs associated with:
| |
• | amortization of deferred financing costs, including the effects of a related party guarantee of our outstanding credit facility; |
| |
• | a loss on the extinguishment of debt; and |
| |
• | fair value adjustments to our derivative liability for warrants issued in conjunction with the related party convertible debt. |
Interest paid on our outstanding bank debt composed substantially all of the remaining other (income) expense.
In April 2010, we entered into a $15.0 million credit facility agreement with HSBC Bank USA, National Association, or HSBC, which we refer to as the 2010 Credit Agreement. This 2010 Credit Agreement was guaranteed by a related party. We concluded that the guarantee represented a deemed contribution and recognized the value of the guarantee as deferred financing costs. The value of the guarantee was determined based on the difference between the 2010 Credit Agreement’s stated interest rate and the interest rate that would apply if there had been no guarantee from the related party. The value was determined to be $6.3 million at the time the 2010 Credit Agreement was established and was amortized over the life of the 2010 Credit Agreement. On March 8, 2013, the 2010 Credit Agreement and related party guarantee were extended through 2014, under an amendment referred to as the 2013 Credit Agreement. At the time of the extension, we concluded that the value of the new guarantee was $3.9 million. This amount was recorded as deferred financing costs and was being amortized through the year 2014.
Upon completion of our IPO on May 7, 2014, the entire outstanding balance of the 2013 Credit Agreement, amounting to $15.0 million plus accrued interest, was paid in full using the proceeds from the IPO. We recorded a loss on the extinguishment of debt of $1.4 million in the three month period ended June 30, 2014, as the remaining deferred financing costs associated with the 2013 Credit Agreement were written off. We had no outstanding debt as of December 31, 2015 and 2014.
We issued common stock warrants in connection with convertible promissory notes and in connection with certain convertible preferred stock agreements that are described in Notes 6 and 8 of our annual audited financial statements in this Form 10-K. All such warrants met the definition of a derivative financial instrument and were accounted for as derivatives. Changes in the combined fair value of the outstanding common stock warrant derivative liabilities were recorded as other (income) expense within the statements of operations in this Form 10-K. All outstanding common stock warrants were exercised in connection with our IPO in May 2014. The combined fair values of the common stock warrant derivative liabilities was $2.7 million as of May 2, 2014, and this amount was reclassified to additional paid-in capital.
The accounting for these transactions are described in Notes 6, 8 and 9 to our audited financial statements included in this Form 10-K.
Income Tax (Expense) Benefit
Income tax (expense) benefit consists of U.S. federal and state income taxes. To date, we have not been required to pay U.S. federal income taxes because of our current and accumulated net operating losses. However, in accordance with U.S. GAAP, for periods in which we reported pre-tax income from discontinued operations for financial reporting purposes and pre-tax loss from continuing operations, we presented income from discontinued operations net of income tax expense attributable to our discontinued operations using the effective tax rate of the Services Business. We also recognized a corresponding income tax benefit on our loss from continuing operations in the corresponding period.
No income tax benefit was recognized during the year ended December 31, 2015, because it is directly correlated to income tax expense in discontinued operations and there was no corresponding income tax expense in discontinued operations in 2015. During the year ended December 31, 2014, we recognized an income tax benefit equal to the corresponding income tax expense on income from discontinued operations for the period.
Discontinued Operations
Discontinued operations comprises revenues, costs, gains and losses directly attributable to our Services Business, which we divested through a sale transaction that closed in July 2015; as summarized below:
| |
• | Revenue included in discontinued operations comprises revenue from the provision of our contract research and development services, which were provided by our Services Business. Our revenue recognition policy is described within Note 2 to our audited financial statements for the year ended December 31, 2015, included in this Form 10-K. |
| |
• | Cost of revenue included in discontinued operations primarily consists of salaries and personnel-related costs, including employee benefits and any stock-based compensation, incurred to generate our contract research and development services revenues. Additional expenses include facilities and equipment costs directly associated with generating revenue, allocated overhead, materials, contracted consultants and other direct costs. We allocate expenses associated with our facilities, information technology costs, and depreciation and amortization, between cost of revenue and operating expenses. Allocations are based on employee headcount or facility square footage utilization, and are determined by the nature of work performed. |
| |
• | Gain on insurance recovery included in discontinued operations in 2014 relates to a reimbursement received from our insurance carrier during the year ended December 31, 2014, for the replacement cost of a fixed asset that was damaged by severe weather. The asset’s net book value was reduced upon occurrence of the damage. The proceeds received from the insurance recovery exceeded the net book value of the asset in the amount of $0.2 million, which we recognized as a gain during the year ended December 31, 2014. This asset was directly associated with our Services Business and, as a result, the gain was included within discontinued operations. |
| |
• | Severance costs included in discontinued operations are exit and disposal costs directly attributable to the sale of the Services Business and incurred pursuant to the Services Business Plan, as described in "Recent Developments" above. |
| |
• | Impairment charge from classification of assets as held for sale included in discontinued operations relates to the carrying value of Services Business property and equipment, net that was in excess of fair value less cost to sell. As described in Note 18 to our audited financial statements in this Form 10-K, we met the relevant criteria for reporting the Services Business as held for sale and in discontinued operations as of June 30, 2015, pursuant to FASB Topic 205-20, Presentation of Financial Statements--Discontinued Operations, and FASB Topic 360, Property, Plant, and Equipment. As a result, we were required to assess the Services Business asset group for impairment pursuant to FASB Topic 360. Our assessment identified an impairment charge of $1.4 million that we recorded in the quarterly period ended June 30, 2015. To determine the impairment charge, pursuant to FASB Topic 360, the net carrying value of the Services Business asset group |
was compared to its fair value as of May 4, 2015. We determined that the selling price paid by Accuratus to acquire the Services Business asset group was the best estimate of fair value. Our valuation methodology is described further in Note 16 of the accompanying audited financial statements in this Form 10-K. We subsequently recorded a $0.1 million loss on disposal in the quarterly period ended September 30, 2015, due to (i) a difference between estimated and final direct selling costs and (ii) a change in estimated working capital of the Services Business between June 30, 2015 and the effective date of the sale on July 17, 2015.
| |
• | Income tax expense included in discontinued operations consists of U.S. federal and state income taxes. To date, we have not been required to pay U.S. federal income taxes because of our current and accumulated net operating losses. However, in accordance with U.S. GAAP, for periods in which we reported pre-tax income from discontinued operations for financial reporting purposes and pre-tax loss from continuing operations, we presented income from discontinued operations net of income tax expense attributable to our discontinued operations using the effective tax rate of the Services Business. We also recognized a corresponding income tax benefit on our loss from continuing operations for the same affected period. |
Results of Operations for the Years Ended December 31, 2015 and 2014
The following table summarizes our results of operations for the years ended December 31, 2015 and 2014, together with the changes in those items in dollars (in thousands) and period-to-period percentage change:
|
| | | | | | | | | | | | | | |
| Years Ended |
| December 31, 2015 | | December 31, 2014 | | Period-to-Period
Change |
Revenue | $ | 257 |
| | $ | 1,256 |
| | $ | (999 | ) | | (79.5 | )% |
Operating expenses: | | | | |
| |
|
Research and development | 16,440 |
| | 8,287 |
| | 8,153 |
| | 98.4 | % |
Selling, general and administrative | 12,166 |
| | 7,616 |
| | 4,550 |
| | 59.7 | % |
Total operating expenses | 28,606 |
| | 15,903 |
| | 12,703 |
| | 79.9 | % |
Loss from operations | (28,349 | ) | | (14,647 | ) | | (13,702 | ) | | 93.5 | % |
Other (income) expense: | | | | |
| |
|
Amortization of deferred financing costs and debt discount | — |
| | 755 |
| | (755 | ) | | (100.0 | )% |
Loss on extinguishment of debt | — |
| | 1,389 |
| | (1,389 | ) | | (100.0 | )% |
Interest (income) expense | (11 | ) | | 48 |
| | (59 | ) | | (122.9 | )% |
Derivative fair value adjustment | — |
| | (10,080 | ) | | 10,080 |
| | (100.0 | )% |
Other expense | — |
| | 10 |
| | (10 | ) | | (100.0 | )% |
Total other (income): | (11 | ) | | (7,878 | ) | | 7,867 |
| | (99.9 | )% |
Loss from continuing operations before taxes | (28,338 | ) | | (6,769 | ) | | (21,569 | ) | | 318.6 | % |
Income tax benefit | — |
| | 1,166 |
| | (1,166 | ) | | (100.0 | )% |
Loss from continuing operations | (28,338 | ) | | (5,603 | ) | | (22,735 | ) | | 405.8 | % |
Income (loss) from discontinued operations, net of tax expense | (4,285 | ) | | 1,369 |
| | (5,654 | ) | | (413.0 | )% |
Net Loss | $ | (32,623 | ) | | $ | (4,234 | ) | | $ | (28,389 | ) | | 670.5 | % |
Revenue. For the year ended December 31, 2015, revenue decreased to $0.3 million compared to $1.3 million of revenue for the year ended December 31, 2014. The decrease of $1.0 million, or 79.5%, was the result of a $1.0 million upfront non-refundable payment received from Waterstone in the fourth quarter of 2014. We recognized the $1.0 million payment as revenue because we satisfied all deliverables associated with the payment prior to December 31, 2014, and have no remaining substantive performance obligations. Our revenue in 2015 is related to the continued amortization of a non-refundable upfront payment received in August 2013 under our collaboration arrangement with R-Pharm that is being recognized over the relationship period.
Research and Development. For the year ended December 31, 2015, research and development expenses increased to $16.4 million from $8.3 million for the year ended December 31, 2014. The increase of $8.2 million, or 98.4%, was primarily the result of a $7.8 million increase in SCY-078 development third-party service expenses and a $0.4 million increase in employee compensation expense. The increase in SCY-078 development third-party service expenses was primarily related to ongoing clinical development activities, including Phase 1 and Phase 2 studies of the oral formulation of SCY-078, the
preclinical and clinical development of an IV formulation of SCY-078, and continued chemistry, manufacturing, and controls (CMC) activities. The increase in employee compensation expense was primarily due to an increase of $0.3 million related to former Services Business personnel devoting more time and effort to SCY-078 development in 2015 (until the Services Business sale in July 2015). When scientific personnel in our former Services Business devoted time to research and development projects, the associated salaries and personnel-related costs for this effort were included in research and development expense, rather than in costs of revenue, which are included within discontinued operations. The former Services Business personnel continued to provide support for SCY-078 development following the July 2015 sale pursuant to the Services Agreement with Accuratus, including $1.6 million for services provided in the year ended December 31, 2015. This expense is included in the SCY-078 development third-party research and development service expense increase described above.
Selling, General and Administrative. For the year ended December 31, 2015, selling, general and administrative expenses increased to $12.2 million from $7.6 million for the year ended December 31, 2014. The increase of $4.6 million, or 59.7%, was primarily the result of a $3.4 million increase in employee compensation expense and a $1.5 million increase in professional services expenses directly associated with our continuing operations as a regulated, publicly traded company, partially offset by a $0.3 million decrease in other administrative expenses. The increase in employee compensation expense was primarily due to an increase in accrued severance and retention compensation costs totaling $1.5 million and an increase in stock compensation expense of $1.9 million. The increase in severance and retention costs was associated with costs incurred pursuant to the Retention Plan, the compensatory plan with Mr. Osborne, and the Separation Agreement with Dr. Ribeill, as described in the "Recent Developments" section above and in Note 15 to the annual audited financial statements in this Form 10-K. The increase in stock compensation expense is related to incremental compensation expense incurred when stock options were modified in the third quarter of 2015 and new option grants awarded in 2015, which are described in further detail in Note 10 to our audited financial statements in this Form 10-K. The decrease in other compensation costs related to a reduction in salary, bonus, and benefits expenses associated with a reduction in selling, general, and administrative personnel headcount following the sale of the Services Business in July 2015.
Amortization of Deferred Financing Costs and Debt Discount. Amortization of deferred financing costs was $0.8 million in the year ended December 31, 2014, which was associated with our 2013 Credit Agreement deferred financing costs. We amortized these deferred financing costs until May 2014, when we repaid the entire outstanding balance of the 2013 Credit Agreement totaling $15.0 million plus accrued interest using the proceeds from the IPO. There was no amortization in the year ended December 31, 2015, because the 2013 Credit Agreement was repaid in full in May 2014.
Loss on Extinguishment of Debt. Loss on extinguishment of debt was $1.4 million in the year ended December 31, 2014. As described in the preceding paragraph, the entire outstanding balance of the 2013 Credit Agreement was repaid in May 2014. The remaining unamortized balance of the deferred financing costs on the debt settlement date of $1.4 million was immediately recognized as a loss on the extinguishment of debt in the year ended December 31, 2014. There was no gain or loss incurred in the year ended December 31, 2015, because the 2013 Credit Agreement was repaid in full in May 2014.
Derivative Fair Value Adjustment. For the year ended December 31, 2015, derivative fair value adjustment was $0.0 million compared to $10.1 million in the year ended December 31, 2014. The derivative fair value adjustment was a gain in the year ended December 31, 2014 and was due to the decrease in the estimated fair value of our common stock, from $47.74 per share as of December 31, 2013, to $10.00 per share as of May 2, 2014. The warrants to purchase common stock accounted for as derivatives were exercised in May 2014 in conjunction with the IPO, and therefore the remaining derivative liability was reclassified to additional paid in capital at that time. Therefore, no gain or loss was incurred during the year ended December 31, 2015.
Income Tax Benefit. For the year ended December 31, 2015, income tax benefit was $0.0 million compared to $1.2 million in the year ended December 31, 2014. No income tax benefit was recognized during the year ended December 31, 2015, because it is directly correlated to income tax expense in discontinued operations and there was no corresponding income tax expense in discontinued operations in 2015. During the year ended December 31, 2014, we recognized an income tax benefit equal to the corresponding income tax expense on income from discontinued operations for the period. The components of the income or loss from discontinued operations in the two periods are described below.
Discontinued Operations. The following table presents revenue, (expenses), gains, and (losses) attributable to discontinued operations for the years ended December 31, 2015 and 2014, together with the changes in those items in dollars (in thousands) and period-to-period percentage change:
|
| | | | | | | | | | | | | | |
| Years Ended |
| December 31, 2015 | | December 31, 2014 | | Period-to-Period
Change |
Total revenue | $ | 7,408 |
| | $ | 17,768 |
| | $ | (10,360 | ) | | (58.3 | )% |
Operating expenses (credits): | | |
| | | | |
Cost of revenue | 7,296 |
| | 15,446 |
| | (8,150 | ) | | (52.8 | )% |
Research and development | 860 |
| | — |
| | 860 |
| | * |
|
Selling, general and administrative | — |
| | (48 | ) | | 48 |
| | (100.0 | )% |
Gain on insurance recovery | — |
| | (165 | ) | | 165 |
| | (100.0 | )% |
Severance and exit costs (Note 15) | 2,114 |
| | — |
| | 2,114 |
| | * |
|
Impairment charge from classification of assets as held for sale (Note 15) | 1,350 |
| | — |
| | 1,350 |
| | * |
|
Loss on disposal, net of associated transaction costs of $764 | 73 |
| | — |
| | 73 |
| | * |
|
Total operating expenses | 11,693 |
| | 15,233 |
| | (3,540 | ) | | (23.2 | )% |
Income (loss) from discontinued operations before income taxes | (4,285 | ) | | 2,535 |
| | (6,820 | ) | | (269.0 | )% |
Income tax expense | — |
| | (1,166 | ) | | 1,166 |
| | (100.0 | )% |
Income (loss) from discontinued operations, net of income tax expense | $ | (4,285 | ) | | $ | 1,369 |
| | $ | (5,654 | ) | | (413.0 | )% |
* Not meaningful
For the year ended December 31, 2015, we incurred a loss from discontinued operations of $4.3 million compared to income from discontinued operations of $1.4 million for the year ended December 31, 2014. The loss from discontinued operations during the year ended December 31, 2015, resulted from revenue of $7.4 million, cost of revenue, research and development, and selling, general, and administrative expenses of the Services Business of $8.2 million, and non-recurring 2015 costs that included severance charges of $2.1 million associated with the termination of employees in connection with the exit and disposal of the Services Business, an impairment charge on classification of assets as held for sale of $1.4 million, and a loss on disposal of $0.1 million. These non-recurring 2015 costs have been described in further detail within the "Recent Developments" section above. The income from discontinued operations in the year ended December 31, 2014, resulted from revenue of $17.8 million, cost of revenue and selling, general, and administrative expenses of the Services Business of $15.4 million, a gain on sale of assets of $0.2 million, and income tax expense of $1.2 million.
The decreases in revenue and costs of revenue in discontinued operations between the two periods occurred because the Services Business was sold early in the third quarter of 2015, on July 17, 2015. Also contributing to the decrease in revenue between the two periods was a decrease in animal health services caused by a reduction in the scope of services provided under our research services agreement with Merial beginning in January 2015, which, when combined with the effect of the sale of the Services Business, resulted in a $5.1 million decrease in revenue under this agreement for the year ended December 31, 2015. The decrease in income tax expense between the two periods related to the change from income from discontinued operations in the 2014 period to loss from discontinued operations in 2015. Income tax expense was only reported for periods in which we reported pre-tax income from discontinued operations and pre-tax loss from continuing operations.
Liquidity and Capital Resources
Sources of Liquidity
Through December 31, 2015, we funded our operations through revenue from the provision of contract research and development services and from net proceeds of debt and equity issuances. Substantially all of our historical revenue has been generated from the provision of our contract research and development services, which were provided by our Services Business that we divested through a sale transaction that closed in July 2015 (see "Recent Developments" above).
As of December 31, 2015, we had cash and cash equivalents of approximately $47.0 million, compared to $32.2 million as of December 31, 2014. The increase in our cash and cash equivalents was primarily due to our April 2015 follow-on public offering, in which we sold an aggregate of 5,376,622 shares of common stock at a public offering price of $7.70 per share. Net
proceeds were approximately $38.0 million, after deducting underwriting discounts and commissions and offering expenses totaling $3.4 million. The cash increase generated by this offering was partially offset by continued research and development costs associated with our lead product candidate, SCY-078.
We have incurred net losses since our inception, including the year ended December 31, 2015. As of December 31, 2015, our accumulated deficit was $150.1 million. We anticipate that we will continue to incur losses for at least the next several years. We expect that our research and development expenses will continue to increase as we continue to execute our research and drug development strategy. We also expect that we will continue to incur selling, general and administrative expenses to support our public reporting company operations. As a result, we will need additional capital to fund our operations, which we may obtain through one or more of equity offerings, debt financings, or other non-dilutive third-party funding (e.g., grants), strategic alliances and licensing or collaboration arrangements. We may offer shares of our common stock pursuant to our Form S-3 shelf registration statement filed with the SEC on October 30, 2015 and declared effective on November 16, 2015, including the related at-the-market facility filed under the Sales Agreement Prospectus on the same date.
On May 7, 2014, we completed our IPO of our common stock pursuant to a registration statement that was declared effective on May 2, 2014. We sold 6,200,000 shares of our common stock at a price of $10.00 per share. As a result of the IPO, we raised a total of $54.6 million in net proceeds after deducting underwriting discounts and commissions of $3.3 million and offering expenses of $4.1 million. A related party that guaranteed our 2013 Credit Agreement invested $15.0 million during the IPO. The 2013 Credit Agreement is described in Note 6 to our audited annual financial statements in this Form 10-K. Costs directly associated with our IPO were capitalized and recorded as deferred offering costs prior to the completion of the IPO. These costs were recorded as a reduction of the proceeds received in arriving at the amount to be recorded in additional paid-in capital. Upon completion of the IPO, all outstanding shares of our preferred stock were converted into 1,691,884 shares of our common stock. In addition, we issued 275,687 shares of common stock in relation to the warrants to purchase our common stock that were exercised.
On May 7, 2014, $15.0 million of the proceeds received from the IPO was used to pay in full the outstanding principal and all accrued interest under the 2013 Credit Agreement, which is described in Note 6 to our audited annual financial statements in this Form 10-K. This payment fully settled our obligations, and released the related party guarantor from all obligations, under and in relation to the 2013 Credit Agreement. There was no outstanding balance under the 2013 Credit Agreement as of December 31, 2014 or 2015.
Cash Flows
The following table sets forth the significant sources and uses of cash for the years ended December 31, 2015 and 2014 (dollars in thousands):
|
| | | | | | | |
| For the Years Ended December 31, |
| 2015 | | 2014 |
| |
Cash and cash equivalents, January 1 | $ | 32,243 |
| | $ | 1,402 |
|
Net cash used in operating activities | (24,545 | ) | | (9,472 | ) |
Net cash provided by (used in) investing activities | 1,586 |
| | (488 | ) |
Net cash provided by financing activities | 37,701 |
| | 40,801 |
|
Net increase in cash and cash equivalents | 14,742 |
| | 30,841 |
|
Cash and cash equivalents, December 31 | $ | 46,985 |
| | $ | 32,243 |
|
Operating Activities
The $15.1 million increase in net cash used in operating activities for the year ended December 31, 2015, as compared to the year ended December 31, 2014, was primarily due to increases in costs associated with SCY-078 research and development efforts and public reporting company operations. We expect that our research and development expenses will continue to increase as we pursue our SCY-078 development efforts described in the "Recent Developments" section above and we expect we will continue to incur selling, general and administrative expenses to support our operations.
Net cash used in operating activities of $24.5 million for the year ended December 31, 2015, primarily consisted of the $32.6 million net loss adjusted for non-cash charges, offset by a net favorable change in operating assets and liabilities of $4.1 million, that included the non-cash component of an impairment charge on classification of assets as held for sale and on disposal of $0.6 million, depreciation of $0.4 million, and stock-based compensation expense of $3.0 million. The non-cash impairment charge is a discrete, non-recurring event and depreciation expense decreased beginning in the third quarter of 2015
because substantially all of that expense is associated with our lease obligations for our Durham, N.C. facility that was transferred as part of the sale of the Services Business. The net favorable change in operating assets and liabilities included an increase in accrued but unpaid severance and retention costs of $2.6 million, an increase in deferred revenue of $1.0 million, and an increase in accounts payable and other accrued expenses of $1.1 million, partially offset by an increase in prepaid expenses and other assets of $0.6 million. The accrued severance and retention costs are related to the Services Business Plan, the Retention Plan, and the resignations of our former chief financial officer and former president, as described further in the "Recent Developments" section above. We expect the majority of these severance and retention accruals to be settled through cash payments occurring during the first half of 2016. The increase in deferred revenue was due to the receipt of advance payments from customers of the Services Business immediately prior to the sale in July 2015. We retained these cash advance payments but transferred the related performance obligations to Accuratus as part of the sale of the Services Business in July 2015. The increase in accounts payable and accrued expenses is associated with the increased SCY-078 research and development activities in 2015, which in turn resulted in increased payment obligations to third-party service providers as of December 31, 2015. The increase in prepaid expenses and other assets is primarily due to (i) a $0.5 million escrow receivable due from Accuratus that we expect to collect in July 2016 and (ii) a $0.2 million increase in the receivable balance due from R-Pharm for reimbursable research and development expenditures. We expect to collect the outstanding receivable due from R-Pharm during the first half of 2016.
Net cash used in operating activities of $24.5 million for the year ended December 31, 2015, includes $2.3 million of net cash used in the operating activities of our Services Business, as reported within discontinued operations, that we do not expect to continue on a prospective basis following the July 2015 sale of the Services Business.
Net cash used in operating activities of $9.5 million for the year ended December 31, 2014, primarily consisted of loss from continuing operations of $5.6 million, adjusted by favorable non-cash charges for a loss on extinguishment of debt of $1.4 million, income from discontinued operations, net of non-cash income tax expense, of $1.4 million, depreciation of $1.2 million, stock-based compensation expense of $1.2 million, the amortization of deferred financing and debt discount costs of $0.8 million, and a favorable change in operating assets and liabilities of $0.6 million. These favorable adjustments were offset by an adjustment for the non-cash gain on the change in fair value of derivative liabilities of $10.1 million in the period, which was described in the "Components of Operating Results" section above and a change in deferred rent of $0.2 million.
Investing Activities
Net cash provided by investing activities of $1.6 million for the year ended December 31, 2015, consisted of $2.5 million of cash proceeds received in July 2015 upon closing of the sale of our Services Business, partially offset by purchases of property and equipment of $0.6 million, a purchase of a security posted as collateral for the Company's corporate credit card program of $0.3 million, and a security deposit payment of $0.1 million for our leased facility in New Jersey. The cash proceeds from the sale were discrete, non-recurring cash flows in the period that we do not expect to occur in future periods. Our cash used for purchases of property and equipment was substantially all related to our Services Business operations. As a result, we expect a decrease in future cash purchases of property and equipment, other than non-recurring capital expenditures to support continuing operations and associated with our relocation to New Jersey.
Net cash used in investing activities of $0.5 million for the year ended December 31, 2014, consisted of purchases of property and equipment of $0.7 million, partially offset by a receipt of $0.2 million in proceeds from an insurance recovery during the second quarter of 2014.
Financing Activities
Net cash provided by financing activities of $37.7 million for the year ended December 31, 2015, consisted of $41.4 million of gross proceeds from our April 2015 follow-on public offering, partially offset by related underwriting discounts and commissions and offering expenses totaling $3.4 million. We also (i) paid offering costs of $0.4 million that were directly associated with our Form S-3 shelf registration statement filed in October 2015 and declared effective by the SEC in November 2015 and (ii) received proceeds from the issuance of shares of our common stock to employees under the terms of our employee stock purchase plan.
Net cash provided by financing activities of $40.8 million for the year ended December 31, 2014, consisted of $62.0 million of gross proceeds received from our IPO in May 2014 and $0.5 million in proceeds raised from the issuance of shares of our D-2 preferred stock in January 2014, offset partially by a $15.0 million payment to settle all outstanding borrowings under our 2013 Credit Agreement and $6.9 million of payments for deferred offering costs and underwriting discounts and commissions. We also received proceeds from (i) the conversion of common stock warrants in connection with our IPO, (ii) the issuance of shares of our common stock to employees under the terms of our employee stock purchase plan and (iii) the exercise of stock options.
Future Funding Requirements
To date, we have not generated any revenue from product sales. We do not know when, or if, we will generate any revenue from product sales. We do not expect to generate significant revenue from product sales unless and until we obtain regulatory approval of and commercialize SCY-078. In addition, we expect our expenses to increase in connection with our ongoing development activities, particularly as we continue the research, development and clinical trials of, and seek regulatory approval for, our product candidates. Although we successfully raised net proceeds of approximately $38.0 million in a follow-on public offering in April 2015, we anticipate that we will need substantial additional funding in connection with our future continuing operations.
As described in the "Recent Developments" section above, we completed the sale of our Services Business pursuant to an Asset Purchase Agreement, effective July 17, 2015, with Accuratus for an aggregate purchase price of $3.9 million, subject to a pre-closing working capital adjustment of $0.8 million. In addition, a portion of the consideration payable at closing equal to $0.5 million was withheld and is subject to an escrow for a period of 12 months from the date of closing to satisfy our indemnification obligations in connection with breaches of any representation and warranties and other customary obligations under the terms of the Agreement. The resulting net proceeds received by us at closing in July 2015 totaled approximately $2.5 million.
Based upon our current operating plan, we believe that our existing cash and cash equivalents will enable us to fund our operating expenses and capital expenditure requirements into the second quarter of 2017. We are currently evaluating our operating plan and assessing the potential cash utilization impact of SCY-078 research and development strategy updates, which are described in the "Recent Developments" section above. We have based our estimates on assumptions that may prove to be wrong, and we may use our available capital resources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development and commercialization of product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures necessary to complete the development of our product candidates.
Our future capital requirements will depend on many factors, including:
| |
• | the progress, costs, and the clinical research and development of SCY-078; |
| |
• | the outcome, costs and timing of seeking and obtaining FDA and any other regulatory approvals; |
| |
• | the ability of our product candidates to progress through clinical development successfully; |
| |
• | our need to expand our research and development activities; |
| |
• | the costs associated with securing, establishing and maintaining commercialization and manufacturing capabilities; |
| |
• | our ability to maintain, expand and defend the scope of our intellectual property portfolio, including the amount and timing of any payments we may be required to make, or that we may receive, in connection with the licensing, filing, prosecution, defense and enforcement of any patents or other intellectual property rights; |
| |
• | our need and ability to hire additional management and scientific and medical personnel; |
| |
• | our need to implement additional, as well as to enhance existing, internal systems and infrastructure, including financial and reporting processes and systems; |
| |
• | the costs associated with capital expenditures needed to support our continuing operations; and |
| |
• | the economic and other terms, timing and success of our existing licensing arrangements and any collaboration, licensing or other arrangements into which we may enter in the future. |
Until such time, if ever, as we can generate substantial revenue from product sales, we expect to finance our cash needs through a combination of net proceeds from equity offerings, debt financings, or other non-dilutive third-party funding (e.g., grants), marketing and distribution arrangements, or other collaborations, strategic alliances or licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, as we did in April 2015, the ownership interests of our common stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of our common stockholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional funds through sales of assets, other non-dilutive third-party funding, marketing and distribution arrangements or other collaborations, strategic alliances or licensing
arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or to grant licenses on terms that may not be favorable to us.
Contractual Obligations, Commitments and Contingencies
Our commitments and contingencies, including payment obligations under license agreements that are contingent upon future events such as our achievement of specified development, regulatory and commercial milestones, have been disclosed in Notes 7, 15, 17 and 18 of our audited financial statements for the year ended December 31, 2015, included in this Form 10-K.
In addition to those obligations, commitments and contingencies set forth in Notes 7, 15, 17 and 18, we have and will continue to enter into contracts in the normal course of business with various third parties who support our clinical trials, support our preclinical research studies, and provide other services related to our operating purposes. These contracts generally provide for termination or cancellation within 30 days of notice.
Off-Balance Sheet Arrangements
During the periods presented we did not have, nor do we currently have, any off-balance sheet arrangements as defined under SEC rules.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which we have prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of our financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of our financial statements, as well as the reported revenues and expenses during the reported periods. We evaluate these estimates and judgments on an ongoing basis. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
While our significant accounting policies are more fully described in Note 2 to our financial statements for the year ended December 31, 2015, included in this annual report, we believe that the following accounting policies are critical to the process of making significant judgments and estimates in the preparation of our financial statements and understanding and evaluating our reported financial results.
Revenue Recognition and Deferred Revenue
We have historically derived substantially all of our revenue from contract research and development services performed under fee for service arrangements, which have been provided by our Services Business that was sold in July 2015. We have also entered into collaboration and licensing agreements in which multiple elements exist, including the sale of licenses and the provision of services, in exchange for non-refundable upfront payments and consideration as services are performed. Under these arrangements, we are also entitled to receive development milestones and royalties in the form of a designated percentage of product sales. We classify non-refundable upfront payments, milestone payments and royalties received under collaboration and licensing agreements as revenues within our statements of operations because we view such activities as being central to our business operations.
We recognize revenue when there is persuasive evidence of an arrangement, delivery has occurred or we have provided the service, the fees are fixed and determinable and collectability is reasonably assured. We record amounts received prior to satisfying the above revenue recognition criteria as deferred revenue until all applicable revenue recognition criteria are met.
Non-refundable upfront fees are recorded as deferred revenue and recognized into revenue as license fees from collaborations on a straight-line basis over the estimated period of our substantive performance obligations. If we do not have substantive performance obligations, we recognize non-refundable upfront fees into revenue through the date the deliverable is satisfied. Analyzing the arrangement to identify deliverables requires the use of judgment, and each deliverable may be an obligation to deliver services, a right or license to use an asset, or another performance obligation.
We will recognize a milestone payment as revenue when earned if it is substantive and we have no ongoing performance obligations related to the milestone. A milestone payment is considered substantive if it: 1) is commensurate with either our performance to achieve the milestone or the enhanced value of the delivered item as a result of a specific outcome from our performance to achieve the milestone; 2) relates solely to past performance; and 3) is reasonable relative to all of the deliverables and payment terms, including other potential milestone consideration, within the arrangement.
We have received several non-refundable upfront payments under certain licensing and collaboration arrangements that contain substantive prospective performance obligations that we are providing to our licensees or collaboration partners over defined or estimated service or relationship periods. Because these arrangements contained substantive performance obligations, the non-refundable upfront payments are being recognized over the service periods of each respective arrangement. Revenue recognized under these non-refundable upfront payments are described further in Note 2 to our audited financial statements for the year ended December 31, 2015, included in this annual report.
In November 2014, we received a $1.0 million non-refundable upfront payment from Waterstone Pharmaceuticals (HK Limited) ("Waterstone") under our license agreement with Waterstone (described in Note 17 to our audited financial statements for the year ended December 31, 2015, included in this Form 10-K). We analyzed the arrangement and concluded we have no remaining substantive obligations to perform under the arrangement after December 31, 2014. As a result, we recognized revenue of $1.0 million from this non-refundable upfront payment during the year ended December 31, 2014. The development milestone payment and the royalties potentially due to us under the arrangement will be recognized as revenue if and when we receive the payments.
Research and Development Accruals
We are required to estimate our expenses resulting from our obligations under contracts with CROs, clinical site agreements, vendors, and consultants in connection with conducting SCY-078 clinical trials and preclinical studies and other development activities. The financial terms of these contracts are subject to negotiations which vary from contract to contract, and may result in payment flows that do not match the periods over which materials or services are provided to us under such contracts. Our objective is to reflect the appropriate development and trial expenses in our financial statements by matching those expenses with the period in which the services and efforts are expended by our service providers.
For clinical trials, we account for these expenses according to the progress of the trial as measured by actual hours expended by CRO personnel, investigator performance or completion of specific tasks, patient progression, or timing of various aspects of the trial. For preclinical development services performed by outside service providers, we determine accrual estimates through financial models, taking into account development progress data received from outside service providers and discussions with our knowledgeable internal personnel and service provider personnel. During the course of a clinical trial or preclinical study or development project, we adjust our rate of trial or project expense recognition if actual results differ from our estimates. We make estimates of our accrued expenses as of each balance sheet date within our financial statements based on the facts and circumstances known to us at that time. Our understanding of the status and timing of services performed relative to the actual status and timing of services performed may vary and may result in our reporting changes in estimates in any particular period. We have not experienced any significant adjustments to our estimates to date.
Stock-Based Compensation
We record the fair value of stock options issued as of the grant date as compensation expense. We recognize compensation expense over the requisite service period, which is equal to the vesting period.
Stock-based compensation expense has been reported in our statements of operations as follows (dollars in thousands):
|
| | | | | | | |
| Years Ended December 31, |
| 2015 | | 2014 |
| |
Research and development | $ | 300 |
| | $ | 394 |
|
Selling, general and administrative | 2,515 |
| | 648 |
|
Discontinued operations | 208 |
| | 159 |
|
Total | $ | 3,023 |
| | $ | 1,201 |
|
On December 31, 2015, the aggregate intrinsic value of outstanding options to purchase shares of our common stock was $0.00, based upon the $6.21 closing sales price per share of our common stock as reported on the NASDAQ Global Market on that date.
Determination of the Fair Value of Stock-based Compensation Grants
We calculate the fair value of stock-based compensation arrangements using the Black-Scholes option-pricing model. The Black-Scholes option-pricing model requires the use of subjective assumptions, including volatility of our common stock, the expected term of our stock options, the risk free interest rate for a period that approximates the expected term of our stock options, and the fair value of the underlying common stock on the date of grant. In applying these assumptions, we considered the following factors:
| |
• | we do not have sufficient history to estimate the volatility of our common stock price. We estimate expected volatility based on reported data for selected reasonably similar publicly traded companies for which the historical information is available. For the purpose of identifying peer companies, we consider characteristics such as industry, length of trading history, similar vesting terms and in-the-money option status. We plan to continue to use the guideline peer group volatility information until the historical volatility of our common stock is relevant to measure expected volatility for future option grants; |
| |
• | the assumed dividend yield is based on our expectation of not paying dividends on our underlying common stock for the foreseeable future; |
| |
• | we determine the average expected life of stock options based on the simplified method in accordance with SEC Staff Accounting Bulletin Nos. 107 and 110, as our common stock has a limited trading history. We expect to use the simplified method until we have sufficient historical exercise data to provide a reasonable basis upon which to estimate expected term; |
| |
• | we determine the risk-free interest rate by reference to implied yields available from U.S. Treasury securities with a remaining term equal to the expected life assumed at the date of grant; and |
| |
• | we estimate forfeitures based on our historical analysis of actual stock option forfeitures. |
As described in Note 10 to our audited financial statements for the year ending December 31, 2015, included in this Form 10-K, we recognized additional compensation expense in connection with certain stock option award term modifications that were approved by our board of directors in June, July and September 2015 and June 2014 and by our shareholders in September 2014. The additional compensation expense was determined in accordance with FASB ASC Topic 718, Compensation--Stock Compensation, and we calculated the incremental fair value of the modified option awards using the Black-Scholes option-pricing model. We considered the same previously described factors when we identified the appropriate assumptions used in the Black-Scholes option-pricing model to determine incremental fair value of the modified option awards.
The assumptions used in the Black-Scholes option-pricing model for the years ended December 31, 2015 and 2014, are set forth below:
|
| | | |
Employee Stock Options | Years Ended
December 31, |
| 2015 |
| 2014 |
Weighted average risk-free interest rate | 1.60% | | 2.05% |
Weighted average expected term (in years) | 6.07 | | 6.04 |
Weighted average expected volatility | 64.42% | | 68.57% |
Expected dividend yield | —% | | —% |
Forfeiture rate | 5.00% | | 5.00% |
|
| | | |
Non-Employee Stock Options | Years Ended
December 31, |
| 2015 | | 2014 |
Weighted average risk-free interest rate | 1.62% | | 1.75% |
Weighted average expected term (in years) | 5.32 | | 5.30 |
Weighted average expected volatility | 63.46% | | 64.10% |
Expected dividend yield | —% | | —% |
Forfeiture rate | 5.00% | | 5.00% |
Determination of the Fair Value of Common Stock on Grant Dates
Historically, we have granted stock options at exercise prices not less than the fair value of our common stock. Prior to our IPO in May 2014, we were a private company with no active public market for our common stock. Therefore, our board of directors estimated per share fair value of our common stock at each grant date using recently prepared valuations performed in accordance with the guidance outlined in the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held Company Equity Securities Issued as Compensation, also known as the Practice Aid. In conducting these valuations, our board of directors considered all objective and subjective factors that it believed to be relevant, including its and management’s best estimate of our business condition, prospects and operating performance at each grant date. In reaching these fair value determinations, our board of directors and management considered a range of objective and subjective factors and assumptions including, among others:
| |
• | our results of operations, financial position, status of our research and development efforts, stage of development and business strategy and the material risks related to our business and industry; |
| |
• | external market conditions affecting the life sciences and biotechnology industry sectors; |
| |
• | the prices at which we sold shares of convertible preferred stock to third-party investors; |
| |
• | the superior rights and preferences of the convertible preferred stock relative to our common stock at the time of each grant; |
| |
• | the valuation of publicly traded companies in the life sciences and biotechnology sectors, as well as recently completed mergers and acquisitions of peer companies; |
| |
• | the lack of an active public market for our common stock and convertible preferred stock; |
| |
• | the likelihood of achieving a liquidity event in light of prevailing market conditions, such as an initial public offering or sale of our company; and |
| |
• | any recent contemporaneous valuations prepared in accordance with methodologies outlined in the Practice Aid. |
Estimating the fair value of our common stock prior to our IPO in May 2014 was highly complex and subjective because our shares were not publicly traded. We used a probability-weighted expected return method, or PWERM, to estimate the fair value of common stock prior to our IPO. Significant inputs for the PWERM included an estimate of our equity value, a weighted average cost of capital, and an estimated probability and timing for each valuation scenario.
For all grants of stock options made following the completion of our IPO, we have determined, and will determine in the future, fair value based on the closing price of our common stock on the Nasdaq Global Market on the date of determination. As a result, the fair value of our common stock no longer requires a highly complex and subjective estimation process.
Fair Value Adjustments to Warrant Liability
We issued warrants to purchase our common stock in connection with the issuances of convertible notes and the issuance of Series D-2 convertible preferred stock. In connection with the consummation of the IPO in May 2014, substantially all outstanding common stock warrants were exercised at an exercise price of $0.20 per share and the holders received 275,687 shares of common stock.
We calculated the fair value of common stock warrants at their intrinsic value, which was the estimated fair value of the common stock less the exercise price for the warrant. We estimated fair value of our common stock using the methodology described in the previous section, titled "Determination of the Fair Value of our Common Stock on Grant Dates." As described in Note 6 to our audited financial statements for the year ending December 31, 2014, included in this Form 10-K, at the date of issuance, the fair value of the warrants issued with convertible notes was recognized as a debt discount to the convertible notes, which was amortized to expense over the stated term of the related notes. As described in Note 8 to our audited financial statements included in this Form 10-K, at the date of issuance, the fair value of the warrants issued with the Series D-2 convertible preferred stock was recognized as a discount to the Series D-2 convertible preferred stock, which was accreted to additional paid-in capital.
As described in Note 9 to our audited financial statements included in this Form 10-K, the warrants issued in connection with both the convertible notes and the convertible preferred stock were also classified as a long-term derivative liability, which was adjusted at each reporting period to reflect its fair value calculated based on the estimated fair value of our common stock. The combined fair value of the common stock warrant derivative liabilities, including warrants issued with the sale of Series D-2 Preferred, was $2.7 million as of May 2, 2014, and this amount was settled to additional paid in capital on that date. The fair value adjustment of the long-term derivative liability for common stock warrants was recorded as other income in the amount of $10.4 million for the year ended December 31, 2014. As discussed in Note 8 to our audited annual financial
statements in this Form 10-K, the fair value of the warrants issued in connection with our Series D-2 Preferred offering in January 2014 was $0.4 million above the face amount of the Series D-2 Preferred. This excess was expensed in the year ended December 31, 2014, and, as a result, the net fair value adjustment presented in the accompanying statements of operations for the year ended December 31, 2014 in this Form 10-K, was income of $10.1 million.
| |
ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK |
As a Smaller Reporting Company until December 31, 2015, we are not required to provide the disclosure required by this Item until our next Annual Report on Form 10-K.
| |
ITEM 8. | FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA |
INDEX TO FINANCIAL STATEMENTS
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders of
SCYNEXIS, Inc.
Jersey City, New Jersey
We have audited the accompanying balance sheets of SCYNEXIS, Inc., (the “Company”) as of December 31, 2015 and 2014, and the related statements of operations, changes in convertible preferred stock and stockholders’ equity, and cash flows for each of the two years in the period ended December 31, 2015. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such financial statements present fairly, in all material respects, the financial position of SCYNEXIS, Inc., as of December 31, 2015 and 2014, and the results of its operations and its cash flows for each of the two years in the period ended December 31, 2015, in conformity with accounting principles generally accepted in the United States of America.
/s/ Deloitte & Touche LLP
Raleigh, North Carolina
March 7, 2016
SCYNEXIS, INC.
BALANCE SHEETS
(in thousands, except share and per share data) |
| | | | | | | |
| December 31, 2015 | | December 31, 2014 |
Assets | | | |
Current assets: | | | |
Cash and cash equivalents | $ | 46,985 |
| | $ | 32,243 |
|
Prepaid expenses and other current assets | 1,452 |
| | 703 |
|
Assets of discontinued operations, net (Note 18) | — |
| | 6,701 |
|
Total current assets | 48,437 |
| | 39,647 |
|
Other assets | 419 |
| | 25 |
|
Deferred offering costs | 417 |
| | — |
|
Total assets | $ | 49,273 |
| | $ | 39,672 |
|
Liabilities and stockholders’ equity | | | |
Current liabilities: | | | |
Accounts payable | $ | 619 |
| | $ | 426 |
|
Accrued expenses | 3,149 |
| | 2,245 |
|
Accrued severance and retention costs (Note 15) | 2,639 |
| | — |
|
Deferred revenue, current portion | 257 |
| | 257 |
|
Liabilities related to assets of discontinued operations (Note 18) | — |
| | 2,420 |
|
Total current liabilities | 6,664 |
| | 5,348 |
|
Deferred revenue, net of current portion | 635 |
| | 893 |
|
Deferred rent | 25 |
| | — |
|
Total liabilities | 7,324 |
| | 6,241 |
|
Commitments and contingencies (Note 7) |
| |
|
Stockholders’ equity: | | | |
Preferred stock, $0.001 par value, authorized 5,000,000 shares as of December 31, 2015 and December 31, 2014; 0 shares issued and outstanding as of December 31, 2015 and December 31, 2014 | — |
|
| — |
|
Common stock, $0.001 par value, authorized 125,000,000 shares as of December 31, 2015, and December 31, 2014; 13,905,599 and 8,512,103 shares issued and outstanding as of December 31, 2015, and December 31, 2014, respectively | 14 |
| | 8 |
|
Additional paid-in capital | 192,069 |
| | 150,934 |
|
Accumulated deficit | (150,134 | ) | | (117,511 | ) |
Total stockholders’ equity | 41,949 |
| | 33,431 |
|
Total liabilities and stockholders’ equity | $ | 49,273 |
| | $ | 39,672 |
|
SCYNEXIS, INC.
STATEMENTS OF OPERATIONS
(in thousands, except share and per share data)
|
| | | | | | | |
| Years Ended December 31, |
| 2015 | | 2014 |
Revenue | $ | 257 |
| | $ | 1,256 |
|
Operating expenses: | | | |
Research and development, net | 16,440 |
| | 8,287 |
|
Selling, general and administrative | 12,166 |
| | 7,616 |
|
Total operating expenses | 28,606 |
| | 15,903 |
|
Loss from operations | (28,349 | ) | | (14,647 | ) |
Other (income) expense: | | | |
Amortization of deferred financing costs and debt discount | — |
| | 755 |
|
Loss on extinguishment of debt | — |
| | 1,389 |
|
Interest (income) expense | (11 | ) | | 48 |
|
Derivative fair value adjustment | — |
| | (10,080 | ) |
Other expense | — |
| | 10 |
|
Total other (income): | (11 | ) | | (7,878 | ) |
Loss from continuing operations before taxes | (28,338 | ) | | (6,769 | ) |
Income tax benefit | — |
| | 1,166 |
|
Loss from continuing operations | (28,338 | ) | | (5,603 | ) |
Income (loss) from discontinued operations, net of tax expense of $0 and $1,166 for the years ended December 31, 2015 and 2014, respectively | (4,285 | ) | | 1,369 |
|
Net loss | $ | (32,623 | ) | | $ | (4,234 | ) |
Deemed dividend for beneficial conversion feature on Series D-2 preferred stock | — |
| | (909 | ) |
Deemed dividend for antidilution adjustments to convertible preferred stock | — |
| | (214 | ) |
Accretion of convertible preferred stock | — |
| | (510 | ) |
Net loss attributable to common stockholders - basic | $ | (32,623 | ) | | $ | (5,867 | ) |
Derivative fair value adjustment | — |
| | (10,080 | ) |
Net loss attributable to common stockholders - diluted | $ | (32,623 | ) | | $ | (15,947 | ) |
Net income (loss) per share attributable to common stockholders - basic | | | |
Continuing operations | $ | (2.33 | ) | | $ | (1.28 | ) |
Discontinued operations | $ | (0.35 | ) | | $ | 0.24 |
|
Net loss per share - basic | $ | (2.68 | ) | | $ | (1.04 | ) |
Net income (loss) per share attributable to common stockholders - diluted | | | |
Continuing operations | $ | (2.33 | ) | | $ | (2.92 | ) |
Discontinued operations | $ | (0.35 | ) | | $ | 0.23 |
|
Net loss per share - diluted | $ | (2.68 | ) | | $ | (2.69 | ) |
Weighted average common shares outstanding: | | | |
Basic | 12,163,559 |
| | 5,663,311 |
|
Diluted | 12,163,559 |
| | 5,937,087 |
|
The accompanying notes are an integral part of the financial statements.
SCYNEXIS, INC.
STATEMENTS OF CHANGES IN CONVERTIBLE PREFERRED STOCK AND STOCKHOLDERS' EQUITY
(in thousands) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Series A Convertible Preferred Stock |
| Series B Convertible Preferred Stock |
| Series C Convertible Preferred Stock |
| Series C-1 Convertible Preferred Stock |
| Series C-2 Convertible Preferred Stock |
| Series D-1 Convertible Preferred Stock |
| Series D-2 Convertible Preferred Stock |
|
| Common Stock |
| Additional Paid-in Capital |
| Accumulated Deficit |
| Total Stockholders’ Equity (Deficit) |
Balances as of December 31, 2013 | $ | 250 |
| | $ | 4,215 |
| | $ | 28,121 |
| | $ | — |
| | $ | 13,500 |
| | $ | 16,952 |
| | $ | 24,119 |
| | | $ | — |
| | $ | 5,168 |
| | $ | (113,277 | ) | | $ | (108,109 | ) |
Net loss | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | — |
| | — |
| | (4,234 | ) | | (4,234 | ) |
Exercise of stock options | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | — |
| | 9 |
| | — |
| | 9 |
|
Stock-based compensation expense (Note 10) | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | — |
| | 1,201 |
| | — |
| | 1,201 |
|
Sale of preferred stock (Note 8) | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 544 |
| | | — |
| | — |
| | — |
| | — |
|
Reclassification of warrants issued with preferred stock to derivative liability (Note 8) | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | (544 | ) | | | — |
| | — |
| | — |
| | — |
|
Beneficial conversion feature for sale of preferred stock (Note 8) | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | 909 |
| | | — |
| | (909 | ) | | — |
| | (909 | ) |
Beneficial conversion feature for antidilution adjustment (Note 8) | — |
| | 18 |
| | 153 |
| | — |
| | 43 |
| | — |
| | — |
| | | — |
| | (214 | ) | | — |
| | (214 | ) |
Adjustment of preferred stock to liquidation value | — |
| | (18 | ) | | (153 | ) | | — |
| | (43 | ) | | — |
| | 724 |
| | | — |
| | (510 | ) | | — |
| | (510 | ) |
Issuance of common stock from the IPO, net of underwriting discounts and commissions and offering expenses (Note 1) | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | 6 |
| | 54,577 |
| | — |
| | 54,583 |
|
Conversion of preferred stock into shares of common stock (Note 8) | (250 | ) | | (4,215 | ) | | (28,121 | ) | | — |
| | (13,500 | ) | | (16,952 | ) | | (25,752 | ) | | | 2 |
| | 88,788 |
| | — |
| | 88,790 |
|
Warrant derivative liability reclassified to additional paid-in capital (Note 9) | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | — |
| | 2,701 |
| | — |
| | 2,701 |
|
Exercise of common stock warrants (Note 9) | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | — |
| | 55 |
| | — |
| | 55 |
|
Issuance of common stock - ESPP | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | — |
| | 68 |
| | — |
| | 68 |
|
Balances as of December 31, 2014 | $ | — |
| | $ | — |
| | $ | — |
| | $ | — |
| | $ | — |
| | $ | — |
| | $ | — |
| | | $ | 8 |
| | $ | 150,934 |
| | $ | (117,511 | ) | | $ | 33,431 |
|
Net loss | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | — |
| | — |
| | (32,623 | ) | | (32,623 | ) |
Stock-based compensation expense (Note 10) | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | — |
| | 3,023 |
| | — |
| | 3,023 |
|
Issuance of common stock from April 2015 Offering, net of underwriting discounts and commissions and offering expenses (Note 1) | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | 6 |
| | 38,006 |
| | — |
| | 38,012 |
|
Issuance of common stock - ESPP | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | — |
| | | — |
| | 106 |
| | — |
| | 106 |
|
Balances as of December 31, 2015 | $ | — |
| | $ | — |
| | $ | — |
| | $ | — |
| | $ | — |
| | $ | — |
| | $ | — |
| | | $ | 14 |
| | $ | 192,069 |
| | $ | (150,134 | ) | | $ | 41,949 |
|
The accompanying notes are an integral part of the financial statements.
SCYNEXIS, INC.
STATEMENTS OF CASH FLOWS
(in thousands) |
| | | | | | | |
| Years Ended December 31, |
| 2015 | | 2014 |
Cash flows from operating activities: | | | |
Net loss | $ | (32,623 | ) | | $ | (4,234 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | |
Non-cash component of impairment loss on classification of assets as held for sale (Note 16) | 586 |
| | — |
|
Gain on insurance recovery | — |
| | (165 | ) |
Loss on disposal of Services Business (Note 18)
| 73 |
| | — |
|
Loss on extinguishment of debt | — |
| | 1,389 |
|
Depreciation | 447 |
| | 1,238 |
|
Stock-based compensation expense | 3,023 |
| | 1,201 |
|
Amortization of deferred financing costs and debt discount | — |
| | 755 |
|
Change in fair value of derivative liability | — |
| | (10,080 | ) |
Changes in deferred rent | (108 | ) | | (187 | ) |
Changes in operating assets and liabilities: | | | |
Accounts receivable and unbilled services | 31 |
| | (439 | ) |
Prepaid expenses, other assets, and deferred costs | (633 | ) | | (490 | ) |
Accounts payable and accrued expenses | 1,066 |
| | 1,575 |
|
Accrued severance and retention cost obligations | 2,639 |
| | — |
|
Deferred revenue | 954 |
| | (35 | ) |
Net cash used in operating activities | (24,545 | ) | | (9,472 | ) |
Cash flows from investing activities: | | | |
Proceeds from insurance recovery | — |
| | 216 |
|
Proceeds from sale of Services Business (Note 18)
| 2,549 |
| | — |
|
Purchase of a security | (300 | ) | | — |
|
Payment of security deposit | (74 | ) | | — |
|
Purchases of property and equipment | (589 | ) | | (704 | ) |
Net cash provided by (used in) investing activities | 1,586 |
| | (488 | ) |
Cash flows from financing activities: | | | |
Proceeds from public offerings | 41,400 |
| | 62,000 |
|
Proceeds from sale of preferred stock | — |
| | 544 |
|
Repayment of debt | — |
| | (15,000 | ) |
Payments of offering costs and underwriting discounts and commissions | (3,805 | ) | | (6,875 | ) |
Proceeds from employee stock purchase plan issuances | 106 |
| | 68 |
|
Proceeds from exercise of stock warrants | — |
| | 55 |
|
Proceeds from exercise of stock options | — |
| | 9 |
|
Net cash provided by financing activities | 37,701 |
| | 40,801 |
|
Net increase in cash and cash equivalents | 14,742 |
| | 30,841 |
|
Cash and cash equivalents, beginning of period | 32,243 |
| | 1,402 |
|
Cash and cash equivalents, end of period | $ | 46,985 |
| | $ | 32,243 |
|
Supplemental cash flow information: | | | |
Cash paid for interest | $ | — |
| | $ | 49 |
|
Noncash financing and investing activities: | | | |
Beneficial conversion feature on sale of Series D-2 preferred stock | $ | — |
| | $ | 909 |
|
Beneficial conversion feature for antidilution adjustment | $ | — |
| | $ | 214 |
|
Adjustment of preferred stock to redemption value | $ | — |
| | $ | 510 |
|
Issuance of warrants allocated to debt discount | $ | — |
| | $ | 906 |
|
Equipment purchases in accounts payable and accrued expenses | $ | — |
| | $ | 34 |
|
Impairment of fixed asset | $ | — |
| | $ | 51 |
|
Deferred offering costs reclassified to additional paid-in capital | $ | 3,388 |
| | $ | 4,126 |
|
Warrant derivative liability reclassified to additional paid-in capital | $ | — |
| | $ | 2,701 |
|
Conversion of convertible preferred stock to common stock | $ | — |
| | $ | 88,790 |
|
Proceeds from sale of Services Business held in escrow (Note 18) | $ | 500 |
| | $ | — |
|
SCYNEXIS, INC.
NOTES TO THE FINANCIAL STATEMENTS
(dollars in thousands, except share and per share data)
| |
1. | Description of Business and Basis of Preparation |
Organization
SCYNEXIS, Inc. (“SCYNEXIS” or the “Company”) is a Delaware corporation formed on November 4, 1999. SCYNEXIS is a pharmaceutical company, headquartered in Jersey City, New Jersey committed to the development and commercialization of novel anti-infectives to address significant unmet therapeutic needs.
Until July 17, 2015, the Company also offered its services in drug discovery and development, primarily in the form of integrated research teams consisting of medicinal, computational, analytical, and process scientists working on a collaborative basis with its customers on research projects. These services were provided by the Company's contract research and development services business (the "Services Business") asset group. As part of the Company's strategic objective to focus its resources on the development of SCY-078, the Company's board of directors directed the Company to divest the Services Business, which was no longer strategic to the Company's business. On July 21, 2015, the Company completed the sale of the Services Business asset group pursuant to an Asset Purchase Agreement (the "Purchase Agreement"), with an effective date of July 17, 2015, with Accuratus Lab Services, Inc. ("Accuratus"), a private-equity backed process chemistry, formulation, manufacturing and analytical development services provider. The material terms of the Services Business sale transaction are described in Note 18.
Discontinued Operations
As described above and more fully in Note 18, the Company completed the sale of the Services Business on July 21, 2015. The accompanying audited financial statements reflect the retrospective adjustments for the periods presented to include the Services Business in discontinued operations pursuant to FASB Topic 205-20, Presentation of Financial Statements--Discontinued Operations, and FASB Topic 360, Property, Plant, and Equipment.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires the Company to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities as of the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates. Significant estimates include: the accounts receivable allowance; the fair value of the Company’s common stock used to measure stock-based compensation for options granted to employees and nonemployees and to determine the fair value of common stock warrants; the Services Business asset group's fair value less costs to sell, which was used to assess the Services Business asset group for impairment; the fair value of convertible preferred stock; the fair value of the Company’s derivative liability; the estimate of services and effort expended by third-party research and development service providers used to recognize research and development expense; and the estimated useful lives of property and equipment.
Reverse Stock-splits
On March 17, 2014, the Company amended its amended and restated certificate of incorporation to implement a 1-for-4 reverse stock split of its common stock. The reverse stock split did not cause an adjustment to the par value or the authorized shares of the common stock. As a result of the reverse stock split, the Company adjusted the share amounts under its employee incentive plans, outstanding options and common stock warrant agreements with third parties.
On April 25, 2014, the Company amended its amended and restated certificate of incorporation to implement an additional 1-for-5.1 reverse stock split of its common stock. The reverse stock split did not cause an adjustment to the par value or the authorized shares of the common stock. As a result of the reverse stock split, the Company further adjusted the share amounts under its employee incentive plans, outstanding options and common stock warrant agreements with third parties.
All disclosures of common shares and per common share data in the accompanying financial statements and related notes reflect these two reverse stock splits for all periods presented.
Initial Public Offering
On May 7, 2014, the Company completed an initial public offering (“IPO”) of its common stock. The Company sold an aggregate of 6,200,000 shares of common stock under the registration statement on Form S-1 declared effective by the SEC on
May 2, 2014, at a public offering price of $10.00 per share. Net proceeds were $54,583, after deducting underwriting discounts and commissions of $3,290 and offering expenses of $4,127. Upon the completion of the IPO, all outstanding shares of the Company’s convertible preferred stock were automatically converted into 1,691,884 shares of common stock and certain outstanding warrants were exercised for an additional 275,687 shares of common stock. In connection with the consummation of the IPO, the Company repaid outstanding debt with a principal balance of $15,000, plus all accrued interest, to the holder of such debt, which was outstanding pursuant to a credit agreement referred to herein as the 2013 Credit Agreement.
April 2015 Follow-On Public Offering
On April 28, 2015, the Company completed a follow-on public offering (the "April 2015 Offering") of its common stock. The Company sold an aggregate of 5,376,622 shares of common stock at a public offering price of $7.70 per share. Net proceeds were approximately $38,012, after deducting underwriting discounts and commissions and offering expenses of approximately $3,388. The significant increase in the shares outstanding beginning in April 2015 has impacted the comparability of the Company's net income (loss) per share calculations between the 2015 and 2014 periods.
Shelf Registration Filing
On October 30, 2015, the Company filed a shelf registration statement on Form S-3 with the SEC which was declared effective on November 16, 2015. The registration statement contained two prospectuses:
| |
• | a base prospectus which covers the offering, issuance and sale by the Company of up to a maximum aggregate offering price of $150,000 of the Company's common stock, preferred stock, debt securities and warrants, including common stock or preferred stock issuable upon conversion of debt securities, common stock issuable upon conversion of preferred stock, or common stock, preferred stock or debt securities issuable upon the exercise of warrants (the "Shelf Registration"), and |
| |
• | a prospectus covering the offering, issuance and sale by the Company of up to a maximum aggregate offering price of $40,000 of the Company's common stock that may be issued and sold under a sales agreement with Cowen and Company, LLC (the "Sales Agreement Prospectus"). |
The common stock that may be offered, issued and sold by the Company under the Sales Agreement Prospectus is included in the $150,000 of securities that may be offered, issued and sold by the Company under the base prospectus. Upon termination of the sales agreement with Cowen and Company, LLC, any portion of the $40,000 included in the Sales Agreement Prospectus that is not sold pursuant to the sales agreement will be available for sale in other offerings pursuant to the base prospectus and a corresponding prospectus supplement, and if no shares are sold under the sales agreement, the full $150,000 of securities may be sold in other offerings pursuant to the base prospectus.
2. Summary of Significant Accounting Policies
Assets of Discontinued Operations
The Company considers assets to be held for sale (i) when management or others having the authority to do so approve a plan to sell the assets, (ii) the assets are available for immediate sale in their present condition, (iii) the Company has initiated an active program to locate a buyer and other actions required to complete the plan to sell the assets, (iv) consummation of the transaction is probable, (v) the assets are being actively marketed for sale at a price that is reasonable in relation to their current fair value, and (vi) the transaction is expected to qualify for recognition as a completed sale, within one year. Following the classification of property and equipment as held for sale, the Company discontinues depreciating the assets and writes down the assets to the lower of carrying value or fair market value, if needed. As described in Note 18, on May 4, 2015, actions taken by the Company's board of directors caused the Company to meet the relevant criteria for reporting the Services Business as held for sale within the accompanying balance sheets and the results of associated operating activities as discontinued operations within the accompanying statements of operations.
Concentration of Credit Risk
Financial instruments, which potentially expose the Company to concentrations of credit risk, consist principally of cash on deposit and cash equivalents held with two banks, which exceed FDIC insured limits, and accounts receivable and unbilled services. Ongoing credit evaluations of customer’s financial condition are performed and independent credit ratings for the associated banks are reviewed by the Company and collateral is not required. The Company's money market fund investment (recognized as cash and cash equivalents) is with what the Company believes to be a high quality issuer. The Company has not experienced any losses in such deposit account.
Revenue recognized from a non-refundable upfront license fee payment received from Waterstone Pharmaceuticals (HK Limited), or Waterstone, a licensing partner (see Note 17), accounted for 0% and 80% of the Company's total revenue in continuing operations for the years ended December 31, 2015 and 2014, respectively. Revenue recognized from a non-
refundable upfront payment from R-Pharm, CJSC (“R-Pharm”), a collaboration partner (see Note 17), accounted for 100% and 20% of the Company's total revenue in continuing operations for the years ended December 31, 2015 and 2014, respectively. No other parties contributed to the Company's total revenue in continuing operations in 2015 or 2014.
One Services Business customer represented 31% and another customer represented 15% of accounts receivable and unbilled services included in assets of discontinued operations, net at December 31, 2014. No other Services Business customer accounted for 10% or more of accounts receivable and unbilled services included in assets of discontinued operations, net.
One Services Business customer, which was a related-party (see Note 13), accounted for 29% and 41% of the Company’s total revenues in discontinued operations for the years ended December 31, 2015 and 2014, respectively. Another customer of the Services Business accounted for 23% and 17% of the Company's total revenues in discontinued operations for the year ended December 31, 2015 and 2014, respectively. No other customer accounted for more than 10% of the Company’s total revenues in discontinued operations in 2015 or 2014.
Cash and Cash Equivalents
The Company considers any highly liquid investments with a remaining maturity of three months or less when purchased to be cash and cash equivalents.
Accounts Receivable and Unbilled Services
Accounts receivable and unbilled services consisted of amounts billed and unbilled under the Company’s Services Business contracts with its Services Business customers included in assets of discontinued operations, net, in the accompanying balance sheets. The Company extended credit to customers without requiring collateral. Accounts receivable were stated at net realizable value. On a periodic basis, the Company evaluated its accounts receivable and established an allowance based on its history of collections and write-offs and the current status of all receivables. The Company did not accrue interest on trade receivables.
Property and Equipment
Property and equipment are stated at cost, less accumulated depreciation. Depreciation is determined on a straight-line basis over the estimated useful lives of the respective assets, which generally range from three to seven years. Leasehold improvements are amortized over the shorter of the useful life of the asset or the term of the related lease. Maintenance and repairs are charged against expense as incurred.
Other Assets
Other assets consist primarily of the refundable long-term deposit on the leased building facility and the restricted cash posted as collateral for the Company's corporate credit card program. These long term deposits and restricted cash outflows are presented as investing activities within the accompanying statements of cash flows.
Deferred Offering Costs
Deferred offering costs are expenses directly related to the IPO, the April 2015 Offering, or the Company's shelf registration statement on Form S-3 filed with the SEC in October 2015 (the "Shelf Registration") (see Note 1). These costs consist of legal, accounting, printing, and filing fees that the Company has capitalized, including fees incurred by the independent registered public accounting firm directly related to the offerings. The IPO deferred offering costs were offset against the IPO proceeds in May 2014 and were reclassified to additional paid-in capital upon completion of the IPO. Deferred costs associated with the April 2015 Offering were offset against the proceeds from the April 2015 Offering and were reclassified to additional paid-in capital upon completion of the April 2015 Offering. Deferred costs associated with the Shelf Registration will be reclassified to additional paid in capital on a pro-rata basis in the event the Company completes an offering under the Shelf Registration, with any remaining deferred offering costs charged to the results of operations at the end of the three-year life of the Shelf Registration. As of December 31, 2015 and 2014, the amount capitalized as deferred offering costs was $417 and $0, respectively.
Impairment of Long-Lived Assets
Long-lived assets to be held and used are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of the asset may not be recoverable. When such events occur, the Company compares the carrying amounts of the assets to their undiscounted expected future cash flows. If the undiscounted cash flows are insufficient to recover the carrying value, an impairment loss is recorded for the difference between the carrying value and fair value of the asset. To date, no such impairment has occurred. During 2015, an impairment charge of $1,350 was recognized within discontinued operations. No other such impairment has occurred.
Revenue Recognition and Deferred Revenue
Historically, the Company has derived the majority of its revenue from providing contract research and development services under fee for service arrangements, which were provided by the Services Business that was sold in July 2015 (see Note 18). The Company also has entered into collaboration arrangements in exchange for non-refundable upfront payments and consideration as services are performed. These arrangements include multiple elements, such as the sale of licenses and the provision of services. Under these arrangements, the Company also is entitled to receive development milestone payments and royalties in the form of a designated percentage of product sales. The Company classifies non-refundable upfront payments, milestone payments and royalties received under collaboration and licensing agreements as revenues within its statements of operations because the Company views such activities as being central to its business operations.
Revenue is recognized when all of the following conditions are met: (i) persuasive evidence of an arrangement exists; (ii) delivery has occurred or services have been rendered; (iii) fees are fixed or determinable; and (iv) collection of fees is reasonably assured. The Company’s contract research and development services revenue is recognized in the period in which the services are performed.
When entering into an arrangement, the Company first determines whether the arrangement includes multiple deliverables and is subject to accounting guidance in ASC subtopic 605-25, Multiple-Element Arrangements. If the Company determines that an arrangement includes multiple elements, it determines whether the arrangement should be divided into separate units of accounting and how the arrangement consideration should be measured and allocated among the separate units of accounting. An element qualifies as a separate unit of accounting when the delivered element has standalone value to the customer. The Company’s arrangements do not include a general right of return relative to delivered elements. Any delivered elements that do not qualify as separate units of accounting are combined with other undelivered elements within the arrangement as a single unit of accounting. If the arrangement constitutes a single combined unit of accounting, the Company determines the revenue recognition method for the combined unit of accounting and recognizes the revenue over the period from inception through the date the last deliverable within the single unit of accounting is delivered.
Non-refundable upfront license fees are recorded as deferred revenue and recognized into revenue on a straight-line basis over the estimated period of the Company’s substantive performance obligations. If the Company does not have substantive performance obligations, the Company recognizes non-refundable upfront fees into revenue through the date the deliverable is satisfied. Analyzing the arrangement to identify deliverables requires the use of judgment and each deliverable may be an obligation to deliver services, a right or license to use an asset, or another performance obligation. In arrangements that include license rights and other non-contingent deliverables, such as participation in a steering committee, these deliverables do not have standalone value because the non-contingent deliverables are dependent on the license rights. That is, the non-contingent deliverables would not have value without the license rights, and only the Company can perform the related services. Upfront license rights and non-contingent deliverables, such as participation in a steering committee, do not have standalone value as they are not sold separately and they cannot be resold. In addition, when non-contingent deliverables are sold with upfront license rights, the license rights do not represent the culmination of a separate earnings process. As such, the Company accounts for the license and the non-contingent deliverables as a single combined unit of accounting. In such instances, the license revenue in the form of non-refundable upfront payments is deferred and recognized over the applicable relationship period, which historically has been the estimated period of the Company’s substantive performance obligations or the period the rights granted are in effect. The Company recognizes contingent event-based payments under license agreements when the payments are received. The Company has not received any royalty payments to date.
The Company will recognize a milestone payment as revenue when earned if it is substantive and the Company has no ongoing performance obligations related to the milestone. A milestone payment is considered substantive if it: 1) is commensurate with either the Company’s performance to achieve the milestone or the enhanced value of the delivered item as a result of a specific outcome from the Company’s performance to achieve the milestone; 2) relates solely to past performance; and 3) is reasonable relative to all of the deliverables and payment terms, including other potential milestone consideration, within the arrangement.
Amounts received prior to satisfying all revenue recognition criteria are recorded as deferred revenue in the accompanying balance sheets.
The Company’s deferred revenue includes non-refundable upfront payments received under certain licensing and collaboration arrangements that contain substantive prospective performance obligations that the Company is providing over respective defined service or estimated relationship periods. Such non-refundable upfront payments are recognized over these defined service or estimated relationship periods. The Company received a non-refundable upfront payment of $1,500 from R-Pharm in August 2013 which is being recognized over a period of 70 months. The Company recognized revenue in continuing
operations from this upfront payment of $257 and $256 for the years ended December 31, 2015 and 2014, respectively. The Company received a non-refundable upfront payment of $500 in January 2014 under a research services agreement supported by the Services Business, which was being recognized over a period of 48 months. The Company recognized revenue in discontinued operations from this upfront payment of $68 and $122 for the years ended December 31, 2015 and 2014, respectively.
Collaboration Arrangements
The Company assesses its contractual arrangements, and presents costs incurred and payments received under contractual arrangements, in accordance with FASB ASC 808, Collaborative Arrangements (Topic 808), when the Company determines that the contractual arrangement incudes a joint operating activity, has active participation by both parties, and both parties are subject to significant risks and rewards under the arrangement. When reimbursement payments are due to the Company under a collaborative arrangement within the scope of Topic 808, the Company determines the appropriate classification for each specific reimbursement payment in the statements of operations by considering (i) the nature of the arrangement, (ii) the nature of the Company’s business operations, and (iii) the contractual terms of the arrangement.
The Company has concluded that the August 2013 development, license, and supply agreement with R-Pharm, combined with the supplemental arrangement in November 2014, is a collaborative arrangement pursuant to Topic 808 and the Company’s previously described accounting policy. This agreement and supplemental arrangement is further described in Note 17. The reimbursements due from R-Pharm for specified research and development costs incurred by the Company are classified as a reduction to research and development expense in the accompanying statements of operations. The reimbursements due to the Company are recorded as a reduction of expense when (i) the reimbursable expenses have been incurred by the Company, (ii) persuasive evidence of a cost reimbursement arrangement exists, (iii) reimbursable costs are fixed or determinable, and (iv) the collection of the reimbursement payment is reasonably assured. Unpaid reimbursement amounts due from R-Pharm at period end are presented as an other current asset in the accompanying balance sheets.
Research and Development
Major components of research and development costs include clinical trial activities and services, including related drug formulation, manufacturing, and other development, preclinical studies, cash compensation, stock-based compensation, fees paid to consultants and other entities that conduct certain research and development activities on the Company’s behalf, materials and supplies, legal services, and regulatory compliance.
The Company is required to estimate its expenses resulting from its obligations under contracts with clinical research organizations, clinical site agreements, vendors, and consultants in connection with conducting SCY-078 clinical trials and preclinical development. The financial terms of these contracts are subject to negotiations which vary from contract to contract, and may result in payment flows that do not match the periods over which materials or services are provided to the Company under such contracts. The Company’s objective is to reflect the appropriate development and trial expenses in its financial statements by matching those expenses with the period in which the services and efforts are expended. For clinical trials, the Company accounts for these expenses according to the progress of the trial as measured by actual hours expended by CRO personnel, investigator performance or completion of specific tasks, patient progression, or timing of various aspects of the trial. For preclinical development services performed by outside service providers, the Company determines accrual estimates through financial models, taking into account development progress data received from outside service providers and discussions with applicable Company and service provider personnel.
Reimbursements of certain research and development costs by parties under collaborative arrangements have been recorded as a reduction of research and development expense presented within the statement of operations. Such reimbursements were made under the collaboration arrangement with R-Pharm, which is further described in Note 17. Information about the Company’s research and development expenses and reimbursements due under collaboration arrangements for the years ended December 31, 2015 and 2014 is presented as follows:
|
| | | | | | | | |
| | Years Ended December 31, |
| | 2015 | | 2014 |
Research and development expense, gross | | $ | 17,380 |
| | $ | 8,513 |
|
Less: Reimbursement of research and development expense | | 940 |
| | 226 |
|
Research and development expense, net of reimbursements | | $ | 16,440 |
| | $ | 8,287 |
|
Patent Expenses
Costs related to filing and pursuing patent applications, as well as costs related to maintaining the Company's existing patent portfolio, are recorded as expense as incurred since recoverability of such expenditures is uncertain.
Fair Value of Financial Instruments
Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date, based on the Company’s principal or, in absence of a principal, most advantageous market for the specific asset or liability.
The Company uses a three-tier fair value hierarchy to classify and disclose all assets and liabilities measured at fair value on a recurring basis, as well as assets and liabilities measured at fair value on a non-recurring basis, in periods subsequent to their initial measurement. The hierarchy requires the Company to use observable inputs when available, and to minimize the use of unobservable inputs when determining fair value. The three tiers are defined as follows:
| |
• | Level 1 — Observable inputs that reflect quoted market prices (unadjusted) for identical assets or liabilities in active markets; |
| |
• | Level 2 — Observable inputs other than quoted prices in active markets that are observable either directly or indirectly in the marketplace for identical or similar assets and liabilities; and |
| |
• | Level 3 — Unobservable inputs that are supported by little or no market data, which require the Company to develop its own assumptions about the assumptions market participants would use in pricing the asset or liability based on the best information available in the circumstances. |
Amortization of Deferred Financing Costs and Debt Discount
Amortization of deferred financing costs and debt discount includes the amortization of debt discount related to the warrants issued with the convertible notes (see Note 6), the amortization of issuance costs related to the convertible notes, and amortization of the deferred financing costs related to a deemed contribution for a guarantee from a related party.
Comprehensive Loss
The Company has no items of comprehensive income or loss other than net loss.
Income Taxes
The Company provides for deferred income taxes under the asset and liability method, whereby deferred income taxes result from temporary differences between the tax bases of assets and liabilities and their reported amounts in the financial statements. Valuation allowances are established when necessary to reduce deferred tax assets to the amount that the Company believes is more likely than not to be realized. The Company recognizes uncertain tax positions when the positions will be more likely than not sustained based solely upon the technical merits of the positions.
The Company applies intraperiod tax allocation guidance pursuant to FASB ASC 740, Income Taxes (Topic 740) to allocate income tax (expense) benefit between pre-tax income (loss) from continuing operations and discontinued operations. For periods in which the Company reports pre-tax income from discontinued operations for financial reporting purposes and pre-tax loss from continuing operations, the Company presents income from discontinued operations net of income tax expense attributable to its discontinued operations using the effective tax rate of the Services Business. The Company also recognizes a corresponding income tax benefit on its loss from continuing operations for the same affected period.
Certain modifications made to an outstanding incentive stock option award at any time after the initial grant dates which are considered to be “material modifications”, as defined within the Internal Revenue Code, may result in the affected award being recharacterized as a non-statutory stock option. The effects of any recharacterization modification for purposes of income tax accounting are recognized on a prospective basis.
Stock-Based Compensation
The Company measures and recognizes compensation expense for all stock-based payment awards made to employees, officers, and directors based on the estimated fair values of the awards as of grant date. The Company values equity instruments and stock options granted to employees and non-employee directors using the Black-Scholes valuation model. The value of the portion of the award that is ultimately expected to vest is recorded as expense over the requisite service periods.
The Company estimated the fair value of common stock warrants granted to lenders at their intrinsic value, which was the estimated fair value of the common stock less the exercise price for the warrant.
Deferred Rent
The Company recognizes rent expense on a straight-line basis over the non-cancelable term of its operating lease and records the difference between cash rent payments and the recognition of rent expense as a deferred rent liability. The Company also records landlord-funded lease incentives, such as reimbursable leasehold improvements, as a deferred rent liability, which is amortized as a reduction of rent expense over the non-cancelable term of its operating lease.
Basic and Diluted Net Loss per Share of Common Stock
The Company uses the two-class method to compute net loss per share because the Company has issued securities, other than common stock, that contractually entitle the holders to participate in dividends and earnings of the Company. The two-class method requires earnings for the period to be allocated between common stock and participating securities based upon their respective rights to receive distributed and undistributed earnings. Holders of each series of the Company’s convertible preferred stock were entitled to participate in dividends, when and if declared by the SCYNEXIS Board of Directors (the "board of directors" or the "board"), that were made to common stockholders, and as a result were considered participating securities.
Under the two-class method, for periods with net income, basic net income per common share is computed by dividing the net income attributable to common stockholders by the weighted average number of shares of common stock outstanding during the period. Net income attributable to common stockholders is computed by subtracting from net income the portion of current year earnings that the participating securities would have been entitled to receive pursuant to their dividend rights had all of the year’s earnings been distributed. No such adjustment to earnings is made during periods with a net loss, as the holders of the participating securities have no obligation to fund losses. Diluted net loss per common share is computed under the two-class method by using the weighted average number of shares of common stock outstanding, plus, for periods with net income attributable to common stockholders, the potential dilutive effects of stock options and warrants. In addition, the Company analyzes the potential dilutive effect of the outstanding participating securities when calculating diluted earnings per share. Under the “treasury stock” method, it is assumed that the warrants and options were exercised at the beginning of the period and that the funds obtained from the exercise were used to reacquire the Company’s common stock at the average market price for the period and includes those securities when they are dilutive. Under the “if-converted” method, it is assumed that the outstanding participating securities convert into common stock at the beginning of the period. The Company reports the more dilutive of the approaches as its diluted net income or net loss per share during the period.
Segment and Geographic Information
Operating segments are defined as components of an enterprise (business activity from which it earns revenue and incurs expenses) about which discrete financial information is available and regularly reviewed by the chief operating decision maker in deciding how to allocate resources and in assessing performance. The Company’s chief operating decision maker (“CODM”) is the Chief Executive Officer. The CODM reviews consolidated operating results to make decisions about allocating resources and assessing performance for the entire Company. The Company views its operations and manages its business as one operating segment. All assets of the Company were held in the United States for the years ended December 31, 2015 and 2014.
Although all operations are based in the United States, the Company generated a portion of its revenue, including revenue in discontinued operations, from customers outside of the United States. All of the Company's revenue from continuing operations was generated from non-refundable upfront payments received under certain licensing and collaboration arrangements with partners located in Russia and China. Information about the Company’s revenue, including revenue in continuing operations and discontinued operations, from different geographic regions for the years ended December 31, 2015 and 2014 is presented as follows:
|
| | | | | | | | | | | | | |
| Years Ended December 31, |
| 2015 | | 2014 |
United States | $ | 6,931 |
| | 90 | % | | $ | 16,422 |
| | 86 | % |
Europe | 477 |
| | 7 | % | | 1,235 |
| | 7 | % |
Other non-US | 257 |
| | 3 | % | | 1,367 |
| | 7 | % |
Total revenue | 7,665 |
| | 100 | % | | 19,024 |
| | 100 | % |
Less: Revenue from discontinued operations | 7,408 |
| | 97 | % | | 17,768 |
| | 93 | % |
Revenue from continuing operations | $ | 257 |
| | 3 | % | | $ | 1,256 |
| | 7 | % |
All sales, including sales outside of the United States, are denominated in United States dollars.
Effect of Recent Accounting Pronouncements
In April 2014, the FASB issued ASU 2014-08, Reporting Discontinued Operations and Disclosures of Disposals of Components of an Entity, or ASU 2014-08. Under ASU 2014-08, only disposals representing a strategic shift in operations that have a major effect on the Company’s operations and financial results should be presented as discontinued operations. Additionally, ASU 2014-08 requires expanded disclosures about discontinued operations that will provide financial statement users with more information about the assets, liabilities, income, and expenses of discontinued operations. The amendments in ASU 2014-08 are effective for fiscal years, and interim periods within those years, beginning after December 15, 2014. The Company adopted this guidance in the first quarter of 2015 and has applied it in the accompanying financial statements for presentation and disclosure of the Services Business as discontinued operations (see Note 18). The Company will also apply, as applicable, the guidance to future dispositions or classifications as held for sale.
In May 2014, the FASB issued ASU No. 2014-09, Revenue from Contracts with Customers: Topic 606, or ASU 2014-09. ASU 2014-09 establishes the principles for recognizing revenue and develops a common revenue standard for U.S. GAAP. The standard outlines a single comprehensive model for entities to use in accounting for revenue arising from contracts with customers and supersedes most current revenue recognition guidance, including industry-specific guidance. In applying the new revenue recognition model to contracts with customers, an entity: (1) identifies the contract(s) with a customer; (2) identifies the performance obligations in the contract(s); (3) determines the transaction price; (4) allocates the transaction price to the performance obligations in the contract(s); and (5) recognizes revenue when (or as) the entity satisfies a performance obligation. The accounting standards update applies to all contracts with customers except those that are within the scope of other topics in the FASB Accounting Standards Codification. The accounting standards update also requires significantly expanded quantitative and qualitative disclosures regarding the nature, amount, timing and uncertainty of revenue and cash flows arising from contracts with customers. This guidance is effective for fiscal years and interim periods within those years beginning after December 15, 2017. The Company is currently evaluating the impact that the implementation of ASU 2014-09 will have on the Company’s financial statements.
In August 2014, the FASB issued ASU No. 2014-15, Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern, or ASU 2014-15. ASU 2014-15 will explicitly require management to assess an entity’s ability to continue as a going concern, and to provide related footnote disclosure in certain circumstances. The new standard will be effective for all entities in the first annual period ending after December 15, 2016. Earlier adoption is permitted. The Company is not early adopting ASU 2014-15. The Company is currently evaluating the impact that the implementation of ASU 2014-15 will have on the Company’s financial statements, and the actual impact will be dependent upon the Company’s liquidity and the nature or significance of future events or conditions that exist upon adopting the updated standard.
In April 2015, the FASB issued ASU No. 2015-03, Simplifying the Presentation of Debt Issuance Costs, or ASU 2015-03. Under ASU 2015-03, the costs of issuing debt will no longer be recorded as an intangible asset, except when incurred before receipt of the funding from the associated debt liability. Rather, debt issuance costs related to a recognized debt liability will be presented on the balance sheet as a direct deduction from the debt liability, similar to the presentation of debt discounts. The costs will continue to be amortized to interest expense using the effective interest method. ASU 2015-03 is effective for fiscal years and interim periods beginning after December 15, 2015, with early adoption permitted. ASU 2015-03 requires retrospective application to all prior periods presented in the financial statements. The Company does not expect that the adoption of ASU 2015-03 will have a material impact on its financial statements.
In April 2015, the FASB issued ASU No. 2015-05, Customer’s Accounting for Fees Paid in a Cloud Computing Arrangement, or ASU 2015-05. ASU 2015-05 provides guidance to entities about whether a cloud computing arrangement includes a software license. Under ASU 2015-05, if a software cloud computing arrangement contains a software license, entities should account for the license element of the arrangement in a manner consistent with the acquisition of other software licenses. If the arrangement does not contain a software license, entities should account for the arrangement as a service contract. ASU 2015-05 also removes the requirement to analogize to ASC 840-10, to determine the asset acquired in a software licensing arrangement. For public companies, ASU 2015-05 is effective for annual periods, including interim periods within those annual periods, beginning after December 15, 2015, and early adoption is permitted. The Company does not expect that the adoption of ASU 2015-05 will have a material impact on its financial statements.
In November 2015, the FASB issued ASU No. 2015-17, Balance Sheet Classification of Deferred Taxes, or ASU 2015-17. ASU 2015-17 provides guidance on balance sheet classification of deferred taxes. The new guidance requires that all deferred tax assets and liabilities, along with any related valuation allowance, be classified as noncurrent on the balance sheet. For public companies, ASU 2015-17 is effective for annual periods, including interim periods within those annual periods, beginning after December 15, 2016, and early adoption is permitted. The Company does not expect that the adoption of ASU 2015-17 will have a material impact on its financial statements.
In February 2016, the FASB issued ASU No. 2016-02, Leases, or ASU 2016-02. The new guidance requires lessees to recognize the assets and liabilities arising from leases on the balance sheet. For public companies, ASU 2016-02 is effective
for annual periods, including interim periods within those annual periods, beginning after December 15, 2018, and early adoption is permitted. The Company does not expect that the adoption of ASU 2016-02 will have a material impact on its financial statements.
3. Prepaid Expenses and Other Current Assets
Prepaid expenses and other current assets consist of the following:
|
| | | | | | | |
| December 31, |
| 2015 | | 2014 |
Prepaid SCY-078 development services | $ | 108 |
| | $ | 109 |
|
Prepaid insurance | 285 |
| | 295 |
|
Other prepaid expenses | 91 |
| | 70 |
|
Other receivable due from R-Pharm | 430 |
| | 226 |
|
Escrow receivable due from Accuratus (Note 18) | 500 |
| | — |
|
Other current assets | 38 |
| | 3 |
|
Total | $ | 1,452 |
| | $ | 703 |
|
Property and equipment consists of the following:
|
| | | | | | | |
| December 31, |
| 2015 | | 2014 |
Equipment | $ | 42 |
| | $ | 8,552 |
|
Furniture and fixtures | — |
| | 375 |
|
Leasehold improvements | — |
| | 13,193 |
|
Total property and equipment | 42 |
| | 22,120 |
|
Less accumulated depreciation | — |
| | 17,285 |
|
Property and equipment, net of accumulated depreciation | 42 |
| | 4,835 |
|
Property and equipment reclassified to assets of discontinued operations, net | — |
| | 4,835 |
|
Property and equipment, net of assets of discontinued operations | $ | 42 |
| | $ | — |
|
Depreciation expense was $447 and $1,238 for the years ended December 31, 2015 and 2014, respectively, including $391 and $1,132 presented in discontinued operations in the accompanying statements of operations for the years ended December 31, 2015 and 2014, respectively. As discussed in Note 18, the Company met the relevant criteria for reporting the Service Business as held for sale on May 4, 2015, and as a result, the Company stopped recording depreciation expense on that date and assessed the property and equipment assets for impairment pursuant to FASB Topic 360, Property, Plant, and Equipment. See Note 16 for the Company's impairment assessment of the Service Business asset group and the resulting impairment charge.
In the quarter ended June 30, 2014, the Company’s insurance carrier remitted proceeds for the replacement cost of a fixed asset that was damaged by severe weather. The asset’s net book value was reduced upon occurrence of the damage. The proceeds received from the insurance recovery exceeded the net book value of the asset by $165, which was recognized as a gain during the quarter ended June 30, 2014. The replacement asset was delivered, installed and placed in service during the quarter ended September 30, 2014. The fixed asset, and the resulting gain, was directly related to the Services Business and, therefore, is presented in discontinued operations in the accompanying statements of operations.
5. Accrued Expenses
Accrued expenses consist of the following:
|
| | | | | | | |
| December 31, |
| 2015 | | 2014 |
Accrued research and development expenses | $ | 1,903 |
| | $ | 293 |
|
Accrued employee bonus compensation | 776 |
| | 1,464 |
|
Employee withholdings | 42 |
| | 156 |
|
Other accrued expenses | 428 |
| | 332 |
|
Total accrued expenses | $ | 3,149 |
| | $ | 2,245 |
|
Severance Costs
In June 2014, the Company reduced its workforce in an effort to reduce operating costs. Employee severance costs associated with this action were $379, which were expensed in the quarter ended June 30, 2014.
In 2015, the Company entered into certain compensatory arrangements and commitments with employees and officers, including severance and retention obligations, the material terms of which are described in Note 15.
Credit Facility Agreement
In April 2010, the Company entered into a $15,000 credit facility agreement with HSBC Bank (the “2010 Credit Agreement”). The agreement comprised a $5,000 term loan and a $10,000 revolving credit facility. Borrowings under the 2010 Credit Agreement carried interest at a rate of London InterBank Offered Rate plus 0.95% per annum. The 2010 Credit Agreement required interest-only payments through March 2013 and was guaranteed by a related party that has an investment in the Company. All outstanding borrowings under the agreement were originally due on March 11, 2013. The 2010 Credit Agreement contained no financial covenants.
At the inception of the 2010 Credit Agreement, a deemed contribution in relation to the guarantee of the 2010 Credit Agreement was recognized as deferred financing costs and amortized over the life of the loan. The value of the guarantee was determined based on the difference between the loan’s stated interest rate and the interest rate that would apply if there had been no guarantee from the related party. The Company determined the value of the 2010 Credit Agreement guarantee to be $6,338, which was amortized over the original life of the loan.
On March 8, 2013, the Company entered into an agreement to amend the 2010 Credit Agreement with HSBC Bank (the “2013 Credit Agreement”). The 2013 Credit Agreement required interest-only payments through December 2014 when all outstanding borrowings were due. Other significant terms of the 2010 Credit Agreement remained the same, which included the guarantee made by a related party that has an investment in the Company. The 2013 Credit Agreement represented a new loan, and the Company determined the value of the extended guarantee under the 2013 Credit Agreement to be $3,930, which was amortized over the term of the 2013 Credit Agreement.
Pursuant to an addendum dated April 29, 2014, upon completion of the IPO on May 7, 2014, the entire outstanding balance of the 2013 Credit Agreement, amounting to $15,000 plus accrued interest, was paid in full using the proceeds from the IPO. The payment on May 7, 2014, released the related party guarantor from all obligations under and in relation to the 2013 Credit Agreement. The Company recorded a loss on the extinguishment of debt of $1,389 in the three month period ended June 30, 2014 as the remaining deferred financing costs associated with the 2013 Credit Agreement were written off. The Company had no outstanding debt as of December 31, 2015.
Amortization of deferred financing costs associated with the 2010 Credit Agreement and 2013 Credit Agreement was $0 and $755 for the years ended December 31, 2015 and 2014, respectively.
The weighted-average interest rate was 0.00% and 1.19% for the years ended December 31, 2015 and 2014, respectively. Interest expense was $0 and $48 for the years ended December 31, 2015 and 2014, respectively.
Note and Warrant Purchase Agreements
In December 2011, the Company executed a Note and Warrant Purchase Agreement (the “December 2011 Note and Warrant Agreement”) to issue convertible notes in an aggregate amount not to exceed $15,000. In 2011 and 2012, under the December 2011 Note and Warrant Agreement, the Company issued convertible notes (the “2011-2012 Notes”) with a total
principal amount of $11,444 to related parties that held investments in the Company. The 2011-2012 Notes included warrants to purchase 26,000 shares of the Company’s common stock at $0.20 per share. The 2011-2012 Notes were convertible into shares of the Company’s stock under various methods as stipulated in the agreement.
In June 2013, the Company executed another Note and Warrant Purchase Agreement (the “June 2013 Note and Warrant Agreement”) with certain existing lenders. Under the June 2013 Note and Warrant Agreement, the lenders agreed to loan to the Company up to $1,500 in exchange for convertible notes (the “June 2013 Notes”). The Company issued June 2013 Notes for an aggregate amount of $899. In addition, the Company agreed to issue warrants to purchase shares of the Company’s common stock upon the request of a majority of the noteholders. The June 2013 Notes were convertible into shares of the Company’s stock using methods described in the agreement. In addition, the June 2013 Notes included conversion of the entire outstanding principal and interest balance into equity securities upon the closing of any equity financing at the option of the noteholders.
On December 11, 2013, the noteholders elected to convert the June 2013 Notes into shares of Series D-2 convertible preferred stock. Also on December 11, 2013, the noteholders elected to convert the 2011-2012 Notes into shares of Series D-1 and Series D-2 convertible preferred stock. There was no outstanding principal or accrued interest associated with the 2011-2012 Notes and June 2013 Notes as of December 31, 2015 and as of December 31, 2014.
| |
7. | Commitments and Contingencies |
Leases
The Company has relocated its corporate headquarters and operating activities to Jersey City, New Jersey and leases its headquarters facilities under a long-term non-cancelable operating lease. On July 13, 2015, the Company entered into a sublease (the "Sublease") that became effective July 22, 2015, to sublet certain premises consisting of 10,141 square feet of space (the "Subleased Premises") located at 101 Hudson Street, Jersey City, New Jersey from Optimer Pharmaceuticals, Inc. The term of the Sublease commenced on August 1, 2015 (the "Commencement Date") and is scheduled to expire on July 30, 2018. No base rent was due under the Sublease until one month after the Commencement Date. Under the Sublease, the Company is obligated to pay monthly base rent of approximately $25 per month, which amount increases by 3% annually on each anniversary of the Commencement Date. In addition, the Company was required to fund a security deposit with the sublandlord in the amount of $74.
Pursuant to the Purchase Agreement, Accuratus assumed the Company’s post-closing obligation under its facility lease in Durham, North Carolina and the Company was released from any and all post-closing liability under the Durham, North Carolina facility operating lease (see Note 18).
Rent expense was approximately $641 and $952 for the years ended December 31, 2015 and 2014, respectively, including $394 and $827 presented in discontinued operations in the accompanying statements of operations for the years ended December 31, 2015 and 2014, respectively. Future minimum lease payments for all operating leases as of December 31, 2015 are as follows:
|
| | | |
| |
2016 | $ | 304 |
|
2017 | 307 |
|
2018 | 182 |
|
2019 | — |
|
2020 | — |
|
Thereafter | — |
|
Total | $ | 793 |
|
License Arrangements with Potential Future Expenditures
As of December 31, 2015, the Company had a license arrangement with Merck Sharp & Dohme Corp., or Merck, as amended, that involves potential future expenditures. Under the license arrangement, the Company exclusively licensed from Merck its rights to SCY-078 in the field of human health. SCY-078 is the Company's lead product candidate. Pursuant to the terms of the license agreement, Merck is eligible to receive milestone payments from the Company that could total $19,000 upon occurrence of specific events, including initiation of a phase 3 clinical study, new drug application, and marketing
approvals in each of the U.S., major European markets and Japan. In addition, Merck is eligible to receive tiered royalties from the Company based on a percentage of worldwide net sales of SCY-078. The aggregate royalties are mid- to high-single digits.
In December 2014, the Company and Merck entered into an amendment to the license agreement that defers the remittance of a milestone payment due to Merck, such that no amount will be due upon initiation of the first phase 2 clinical trial of a product containing the SCY-078 compound (the "Deferred Milestone"). The amendment also increases, in an amount equal to the Deferred Milestone, the milestone payment that will be due upon initiation of the first Phase 3 clinical trial of a product containing the SCY-078 compound. Except as described above, all other terms and provisions of the license agreement remain in full force and effect.
The Company has two additional licensing agreements for other compounds that could require it to make payments of up to $2,300 upon achievement of certain milestones by the Company.
Clinical Development Arrangement
The Company has entered into, and expects to continue to enter into, contracts in the normal course of business with various third parties who support its clinical trials, preclinical research studies, and other services related to its development activities. The scope of the services under these agreements can generally be modified at any time, and the agreement can be terminated by either party after a period of notice and receipt of written notice.
Other Arrangements
The Company entered into an agreement with a third party firm to assist the Company in exploring the divestiture of its Services Business (see Note 18). Pursuant to the terms of the agreement, in the event that the Company was able to complete a divestiture of its Services Business to a third-party, the Company was obligated to pay a success fee to the third party firm for the greater of $500 or 4% of the transaction consideration. As described in Note 18, the Company completed the sale of the Services Business pursuant to the Purchase Agreement, dated July 17, 2015. The Company paid and expensed an initial retainer of $50 prior to the closing of the Service Business sale transaction. In July 2015, the Company paid the $450 remaining success fee to the third-party firm in connection with the closing of the sale transaction.
Certain of the Company's employees continued to operate from the Durham facility immediately after the closing for a period of up to six months pursuant to a facility lease agreement between the Company and Accuratus dated July 17, 2015. Under the facility lease agreement, the Company was obligated to pay a monthly license fee of approximately $8 per month. In addition, under a Transition Services Agreement, Accuratus provided accounting, IT, payroll, personnel and human resources support, and equity compensation plan administration support services to the Company at rates ranging from one hundred to two hundred dollars per hour for a period of time not to extend beyond December 31, 2015.
In connection with the sale of the Services Business, the Company and Accuratus also entered into a Commitment to Services Agreement (the "Services Agreement") pursuant to which Accuratus will provide the Company with certain contract research and development services. The material terms of the Services Agreement are described in Note 18.
Compensatory Arrangements with Employees and Officers
The Company has entered into certain compensatory arrangements and commitments with employees and officers, the material terms of which are described in Note 15.
Preferred Stock
On May 7, 2014, the Company amended and restated its articles of incorporation relating to its approved capital structure. The Company's board of directors has authorized the Company, subject to limitations prescribed by Delaware law, to issue up to 5,000,000 shares of preferred stock with a par value of $0.001 per share in one or more series, to establish from time to time the number of shares to be included in each series and to fix the designation, powers, preferences and rights of the shares of each series and any of its qualifications, limitations or restrictions. The Company's board of directors can also increase or decrease the number of shares of any series of preferred stock, but not below the number of shares of that series then outstanding, without any further vote or action by the our stockholders. The Company's board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of the common stock. There were no shares of preferred stock issued and outstanding as of December 31, 2015 and 2014, respectively.
Convertible Preferred Stock
The Company issued multiple series of convertible preferred stock between 2000 and January 2014. In March 2014, the Company amended its amended and restated certificate of incorporation to require the automatic conversion of all series of
convertible preferred stock into common stock upon the completion of a public offering of common stock with gross proceeds of at least $20,000. In May 2014, upon completion of the IPO, all outstanding shares of convertible preferred stock were converted into an aggregate of 1,691,884 shares of common stock at their respective conversion prices.
Warrants Associated with Convertible Preferred Stock Issuances
In July 2006, the Company issued warrants to purchase 196,923 shares of Series C-1 Preferred Stock, which converted into the right to purchase 14,033 shares of common stock in connection with our IPO, however, we refer to these warrants as our Series C-1 Preferred warrants. The Series C-1 Preferred warrants were issued in conjunction with a loan financing agreement with an original exercise price of $3.25 per share of Series C-1 Preferred, which converted into an exercise price of$45.61 per share of common stock in connection with our IPO. These warrants remain outstanding as of December 31, 2015 and will expire on May 7, 2019, which is the five year anniversary of the Company's IPO. The fair value at the date of grant for these instruments was $459, which was recorded as a debt discount. The debt discount related to these warrants was fully amortized as of December 31, 2010. The Company determined that the warrants should be recorded as a derivative liability and stated at fair value at each reporting period. The Company recorded other income associated with the fair value adjustment for these warrants of $0 and $37 for the years ended December 31, 2015 and 2014, respectively.
The December 11, 2013 Series D-2 Purchase Agreement included warrants to purchase 87,532 shares of the Company’s common stock at $0.20 per share. The fair value of the warrants on the date of issuance was $4,214, which was recorded as a discount to the Series D-2 Preferred. The fair value of the warrants was $1,714 above the face amount of the Series D-2 Preferred and this excess was expensed to derivative fair value adjustment at issuance. As described in Note 9, the warrants were classified as a derivative liability and were stated at fair value at each reporting period end date prior to being exercised in May 2014 in conjunction with the Company’s IPO.
On January 31, 2014, the Company sold 388,641 shares of Series D-2 Preferred to related parties under the Series D-2 Purchase Agreement at $1.40 per share, for an aggregate price of $544. The sale also included warrants to purchase 19,048 shares of the Company’s common stock at $0.20 per share. The fair value of the warrants on the date of issuance was $906. The fair value of the warrants was $362 above the face amount of the Series D-2 Preferred and this excess was expensed to derivative fair value adjustment at issuance. As described in Note 9, the warrants were classified as a derivative liability and were stated at fair value at each reporting period end date prior to being exercised in May 2014 in conjunction with the Company’s IPO.
Authorized, Issued, and Outstanding Common Shares
The Company’s common stock has a par value of $0.001 per share and consists of 125,000,000 authorized shares as of December 31, 2015 and 2014, respectively; 13,905,599 and 8,512,103 shares were issued and outstanding as of December 31, 2015, and December 31, 2014, respectively. The following table summarizes common stock share activity for the years ended December 31, 2015 and 2014:
|
| | |
| Shares of
Common Stock |
Balance, December 31, 2013 | 334,068 |
|
Exercise of stock options | 416 |
|
Conversion of preferred stock | 1,691,884 |
|
Exercise of common stock warrants | 275,687 |
|
Common stock issued through IPO | 6,200,000 |
|
Common stock issued through employee stock purchase plan | 10,048 |
|
Balance, December 31, 2014 | 8,512,103 |
|
Common stock issued through April 2015 Offering | 5,376,622 |
|
Common stock issued through employee stock purchase plan | 16,874 |
|
Balance, December 31, 2015 | 13,905,599 |
|
Shares Reserved for Future Issuance
The Company had reserved shares of common stock for future issuance as follows:
|
| | | | | |
| December 31, |
| 2015 | | 2014 |
Outstanding stock options | 1,379,727 |
| | 615,322 |
|
Outstanding Series C-1 Preferred warrants | 14,033 |
| | 14,033 |
|
For possible future issuance under 2014 Equity Incentive Plan (Note 10) | 552,415 |
| | 180,610 |
|
For possible future issuance under 2014 Employee Stock Purchase Plan (Note 10) | 50,283 |
| | 37,746 |
|
For possible future issuance under 2015 Inducement Plan (Note 10) | 165,000 |
| | — |
|
Total common shares reserved for future issuance | 2,161,458 |
| | 847,711 |
|
Liquidation Rights
In the event of any liquidation or dissolution of the Company, the holders of the common stock are entitled to the remaining assets of the Company legally available for distribution.
Dividends and Voting Rights
The holders of the common stock are entitled to receive dividends if and when declared by the Company. The holders of the common stock have the right to one vote per share.
Common Stock Warrants
The Company had outstanding common stock warrants issued in connection with the Note and Warrant Purchase Agreements (Note 6) and in connection with certain convertible preferred stock agreements (Note 8).
The December 2011 Note and Warrant Purchase Agreement included warrants to purchase 26,000 shares of the Company’s common stock at $0.20 per share. The warrants could be exercised for shares of common stock, in accordance with their terms. The number of shares of common stock that could be purchased by exercising the warrants would vary based on the event that occurred and would be calculated in accordance with the December 2011 Note and Warrant Purchase Agreements (Note 6).
On December 11, 2013, holders of the June 2013 Notes exercised their rights under the June 2013 Note and Warrant Agreement to receive warrants to purchase shares of the Company’s common stock. As a result of this exercise, the Company issued warrants to purchase 88,987 shares of the Company’s common stock to the holders of the June 2013 Notes at an exercise price of $0.20 per share. These warrants were exercisable until June 28, 2018, and would terminate unless exercised prior to an IPO.
On December 11, 2013, in connection with the first Series D-2 Preferred offering, the Company issued warrants to purchase 87,532 shares of the Company’s common stock at an exercise price of $0.20 per share. These warrants were exercisable until December 11, 2018, and would terminate unless exercised prior to an IPO. In addition, as a result of the conversion of the principal and interest outstanding on the 2011-2012 Notes into Series D-1 Preferred and Series D-2 Preferred (Note 6), in accordance with the amended terms of the agreement, the number of common shares underlying the warrants issued in connection with the 2011-2012 Notes was increased by 54,120 to a total of 80,120.
On January 31, 2014, in connection with the second Series D-2 Preferred offering, the Company issued warrants to purchase 19,048 shares of the Company’s common stock at $0.20 per share.
In connection with the consummation of the IPO in May 2014, substantially all outstanding common stock warrants were exercised at an exercise price of $0.20 per share and the holders received 275,687 shares of common stock.
All previously described warrants met the definition of a derivative financial instrument and were accounted for as derivatives. The combined fair value of the common stock warrant derivative liabilities, including warrants issued with the sale of Series D-2 Preferred, was $2,701 as of May 2, 2014, and this amount was settled to additional paid in capital on that date. The fair value adjustment of the long-term derivative liability for common stock warrants was recorded as other income in the amount of $10,442 for the year ended December 31, 2014. As discussed in Note 8, the fair value of the warrants issued in connection with the Company’s Series D-2 Preferred offering in January 2014 was $362 above the face amount of the Series D-2 Preferred. This excess was expensed in the year ended December 31, 2014, and, as a result, the net fair value adjustment presented in the accompanying statements of operations for the year ended December 31, 2014, was income of $10,080.
| |
10. | Stock-based Compensation |
2009 Stock Option Plan
The Company had a share-based compensation plan (the “2009 Stock Option Plan”) under which the Company granted options to purchase shares of common stock to employees, directors, and consultants as either incentive stock options or nonqualified stock options. Incentive stock options could be granted with exercise prices not less than 100% to 110% of the fair market value of the common stock. Options granted under the plan generally vest over three to four years and expire in 10 years from the date of grant.
2014 Equity Incentive Plan
In February 2014, the Company’s board of directors adopted the 2014 Equity Incentive Plan, or the 2014 Plan, which was subsequently ratified by its stockholders and became effective on May 2, 2014 (the “Effective Date”). The 2014 Plan is the successor to and continuation of the 2009 Stock Option Plan. As of the Effective Date, no additional awards will be granted under the 2009 Stock Option Plan, but all stock awards granted under the 2009 Stock Option Plan prior to the Effective Date will remain subject to the terms of the 2009 Stock Option Plan. All awards granted on and after the Effective Date will be subject to the terms of the 2014 Plan. The 2014 Plan provides for the grant of the following awards: (i) incentive stock options, (ii) nonstatutory stock options, (iii) stock appreciation rights, (iv) restricted stock awards, (v) restricted stock unit awards, and (vi) other stock awards. Employees, directors, and consultants are eligible to receive awards.
Under the 2014 Plan, after giving effect to the increases to the share reserve approved by the Company’s stockholders in September 2014, and June 2015, discussed below, the aggregate number of shares of common stock that could be issued from and after the Effective Date (the “share reserve”) could not exceed the sum of (i) 1,122,731 new shares, (ii) the shares that represented the 2009 Stock Option Plan’s available reserve on the Effective Date, and (iii) any returning shares from the 2009 Stock Option Plan. Under the 2014 Plan, the share reserve will automatically increase on January 1st of each year, for a period of not more than 10 years, commencing on January 1, 2015 and ending on January 1, 2024, in an amount equal to 4.0% of the total number of shares of capital stock outstanding on December 31st of the preceding calendar year. The board of directors may act prior to January 1st of a given year to provide that there will be no increase in the share reserve or that the increase will be a lesser number of shares than would otherwise occur.
On June 18, 2014, the Company’s board of directors and compensation committee approved an amendment of the 2014 Plan, subject to stockholder approval, to increase the aggregate number of shares of the Company’s common stock that may be issued under the 2014 Plan by an additional 351,653 shares. All other material terms of the 2014 Plan remained unchanged. The Company’s stockholders approved the 2014 Plan amendment on September 11, 2014.
Pursuant to the terms of the 2014 Plan, on January 1, 2015, the Company automatically added 340,484 shares to the total number shares of common stock available for future issuance under the 2014 Plan.
On February 25, 2015, the Company's board of directors approved an amendment of the 2014 Plan, subject to stockholder approval, to increase the aggregate number of shares of common stock that may be issued pursuant to awards under the 2014 Plan by an additional 510,726 shares. The Company's stockholders approved the 2014 Plan amendment on June 4, 2015. All other material terms of the 2014 Plan otherwise remain unchanged.
As of December 31, 2015, there were 552,415 shares of common stock available for future issuance under the 2014 Plan.
See Note 19 for certain events occurring after December 31, 2015 that affected the number of shares of common stock available for future issuance under the 2014 Plan.
2015 Inducement Plan
On March 26, 2015, the Company's board of directors adopted the 2015 Inducement Plan, or the 2015 Plan. The 2015 Plan provides for the grant of nonstatutory stock options, stock appreciation rights, restricted stock awards, restricted stock unit awards, and other forms of equity compensation (collectively, stock awards), all of which may be granted to persons not previously employees or directors of the Company, or following a bona fide period of non-employment, as an inducement material to the individuals’ entering into employment with the Company within the meaning of NASDAQ Listing Rule 5635(c)(4). The 2015 Plan has a share reserve covering 450,000 shares of common stock. During the year ended December 31, 2015, the Company granted options to purchase 285,000 shares of the Company's common stock under to the 2015 Inducement Plan. As of December 31, 2015, there were 165,000 shares of common stock available for future issuance under the 2015 Plan.
Option Valuation Method
The fair value of a stock option is estimated using an option-pricing model that takes into account as of the grant date the exercise price and expected life of the option, the current price of the underlying stock and its expected volatility, expected dividends on the stock, and the risk-free interest rate for the expected term of the option. The Company has used the simplified method in calculating the expected term of all option grants based on the vesting period. Compensation costs related to share-based payment transactions are recognized in the financial statements upon satisfaction of the requisite service or vesting requirements. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. The Company based its estimated forfeiture rate on historical forfeitures of all stock option grants.
The Company has elected to use the Black-Scholes option-pricing model. The Black-Scholes option-pricing model was developed for use in estimating the fair value of traded options that have no vesting restrictions and are fully transferable rather than for use in estimating the fair value of stock options subject to vesting and transferability restrictions. Using the Black-Scholes option-pricing model, the weighted-average fair value of options granted during 2015 and 2014 was $4.79 and $6.24 per option, respectively. The aggregate fair value of options granted during 2015 and 2014 was $4,217 and $3,249, respectively. The assumptions used to estimate fair value and the resulting grant date fair values are as follows:
|
| | | | | | | |
| Employees | | Nonemployees |
| Years Ended December 31, | | Years Ended December 31, |
| 2015 | | 2014 | | 2015 | | 2014 |
Expected dividend yield | — | | — | | — | | — |
Weighted average expected volatility | 64.42% | | 68.57% | | 63.46% | | 64.10% |
Weighted average risk-free interest rate | 1.60% | | 2.05% | | 1.62% | | 1.75% |
Weighted average expected term (in years) | 6.07 | | 6.04 | | 5.32 | | 5.30 |
Forfeiture rate | 5.00% | | 5.00% | | 5.00% | | 5.00% |
The activity for the 2009 Plan, 2014 Plan and 2015 Plan for the years ended December 31, 2015 and 2014 is summarized as follows:
|
| | | | | | | | | | | | |
| Number of Shares | | Weighted- Average Exercise Price | | Weighted- Average Remaining Contractual Life (in years) | | Aggregate Intrinsic Value |
Outstanding — January 1, 2014 | 137,610 |
| | $ | 25.86 |
| | 5.23 | | $ | 3,097 |
|
Granted | 520,887 |
| | 9.53 |
| (1) | | | |
Exercised | (416 | ) | | 20.40 |
| | | | |
Canceled | (42,759 | ) | | 9.57 |
| | | | |
Outstanding — December 31, 2014 | 615,322 |
| | $ | 9.55 |
| | 9.48 | | $ | 265 |
|
Exercisable — December 31, 2014 | 192,916 |
| | $ | 9.44 |
| | 9.48 | | $ | 88 |
|
Vested or expected to vest — December 31, 2014 | 572,926 |
| | $ | 9.55 |
| | 9.48 | | $ | 247 |
|
Outstanding — January 1, 2015 | 615,322 |
| | $ | 9.55 |
| | 9.48 | | $ | 265 |
|
Granted | 880,116 |
| | $ | 8.17 |
| | | | |
Exercised | — |
| | $ | — |
| | | | |
Canceled | (115,711 | ) | | $ | 9.05 |
| | | | |
Outstanding — December 31, 2015 | 1,379,727 |
| | $ | 8.71 |
| | 7.18 | | $ | — |
|
Exercisable — December 31, 2015 | 635,548 |
| | $ | 9.41 |
| | 4.56 | | $ | — |
|
Vested or expected to vest —December 31, 2015 | 1,342,518 |
| | $ | 8.73 |
| | 7.11 | | $ | — |
|
(1) The weighted average exercise price table takes into consideration the effect of the option award modifications approved by the Company's board of directors on June 18, 2014, and approved by the Company's shareholders on September 11, 2014. These option award modifications are described in further detail below.
The intrinsic values in the table above represent the total intrinsic value (the difference between the Company’s closing stock price as of December 31, 2015 and 2014, and the exercise price multiplied by the number of options).
Information as of December 31, 2015, concerning currently outstanding and vested options is as follows:
|
| | | | | | | | | | |
| | Outstanding | | Exercisable |
Exercise Price | | Number of Shares | | Weighted- Average Remaining Contractual Life (in years) | | Number of Shares | | Weighted- Average Remaining Contractual Life (in years) |
$6.53 | | 100,000 |
| | 9.84 | | — |
| | 0.00 |
$6.63 | | 21,600 |
| | 9.95 | | — |
| | 0.00 |
$6.64 | | 66,563 |
| | 9.76 | | 6,563 |
| | 9.76 |
$6.77 | | 2,166 |
| | 0.05 | | 2,166 |
| | 0.05 |
$6.81 | | 17,400 |
| | 9.95 | | — |
| | 0.00 |
$6.89 | | 23,400 |
| | 9.74 | | — |
| | 0.00 |
$7.70 | | 5,944 |
| | 4.88 | | 5,944 |
| | 4.88 |
$8.03 | | 5,700 |
| | 4.80 | | 5,700 |
| | 4.80 |
$8.63 | | 26,114 |
| | 4.79 | | 26,114 |
| | 4.79 |
$8.64 | | 8,800 |
| | 9.56 | | — |
| | 0.00 |
$8.65 | | 169,000 |
| | 9.13 | | 8,800 |
| | 3.73 |
$8.73 | | 9,000 |
| | 3.12 | | 4,000 |
| | 2.66 |
$8.76 | | 429,198 |
| | 8.05 | | 93,454 |
| | 3.84 |
$8.86 | | 4,941 |
| | 9.50 | | 4,941 |
| | 9.50 |
$9.64 | | 455,069 |
| | 4.43 | | 450,864 |
| | 4.39 |
$9.96 | | 9,326 |
| | 7.02 | | 9,326 |
| | 7.02 |
$10.00 | | 8,316 |
| | 9.16 | | 486 |
| | 9.16 |
$10.81 | | 17,190 |
| | 8.93 | | 17,190 |
| | 8.93 |
Total | | 1,379,727 |
| | Total | | 635,548 |
| |
|
The total fair value of shares vested during the years ended December 31, 2015 and 2014 was $595 and $458, respectively, which exclude the effects of the 2015 option amendments (described in further detail below) that resulted accelerated vesting and related incremental fair value.
Unvested shares as of December 31, 2015 and 2014 are as follows:
|
| | | | | | | | |
As of December 31, 2015 | | As of December 31, 2014 |
Exercise Price | | Number of Unvested Shares | | Exercise Price | | Number of Unvested Shares |
$6.53 | | 100,000 |
| | $6.53 | | — |
|
$6.63 | | 21,600 |
| | $6.63 | | — |
|
$6.64 | | 60,000 |
| | $6.64 | | — |
|
$6.77 | | — |
| | $6.77 | | 8,000 |
|
$6.81 | | 17,400 |
| | $6.81 | | — |
|
$6.89 | | 23,400 |
| | $6.89 | | — |
|
$8.64 | | 8,800 |
| | $8.64 | | — |
|
$8.65 | | 160,200 |
| | $8.65 | | — |
|
$8.73 | | 5,000 |
| | $8.73 | | 31,620 |
|
$8.76 | | 335,744 |
| | $8.76 | | — |
|
$9.64 | | 4,205 |
| | $9.64 | | 367,127 |
|
$10.00 | | 7,830 |
| | $10.00 | | — |
|
$10.81 | | — |
| | $10.81 | | 15,660 |
|
Total | | 744,179 |
| | Total | | 422,407 |
|
As of December 31, 2015 and 2014, there was approximately $2,867 and $2,683, respectively, of total unrecognized compensation cost related to unvested share-based compensation arrangements granted under the plan. That cost is expected to be recognized over weighted-average periods of 3.1 and 3.2 years for the years ended December 31, 2015 and 2014, respectively. The aggregate intrinsic value of options exercised during the year ended December 31, 2014 was $11.
2015 Option Amendments
During the year ended December 31, 2015, the following events resulted in the amendment to terms of outstanding stock option awards:
| |
• | On June 4, 2015, the Company's board of directors approved an extension to the existing 90-day post-employment option exercise period to a period ranging from 36 to 48 months for three directors who resigned from the board effective June 4, 2015. The directors held outstanding options to purchase 48,283 shares of the Company's common stock at a weighted average exercise price of $9.01 per share. All outstanding options were fully vested prior to June 4, 2015. |
| |
• | In connection with the Company's sale of its Services Business (see Note 18), the Company designed a compensatory plan to promote the retention of services of non-executive employees supporting that business (the "Services Business Plan"). The complete terms of the Service Business Plan are described in Note 15. The Company terminated certain employees in June 2015 (the "June 2015 Terminated Employees") who became eligible for severance benefits pursuant to the terms of the Services Business Plan. The outstanding stock options held by the June 2015 Terminated Employees were modified to provide: (i) accelerated vesting of all unvested stock options as of the termination date and (ii) an extension to the existing 90-day post-employment option exercise period, which varies for each employee based upon years of service, with a maximum exercise period of 48 months. As of June 30, 2015, the June 2015 Terminated Employees held outstanding options to purchase 17,715 shares of the Company's common stock at a weighted average exercise price of $9.64 per share, including aggregate unvested options to purchase 8,331 shares at a weighted average exercise price of $9.64 per share. |
| |
• | As described in Note 15, Charles F. Osborne, Jr., the Company’s former chief financial officer, resigned from the Company effective June 30, 2015. The Company's compensation committee of the board of directors approved the following modifications to Mr. Osborne's outstanding options to purchase the Company's common stock: (i) accelerated vesting of all unvested stock options as of June 30, 2015, and (ii) an extension to the existing 90-day post-employment option exercise period to 36 months. As of June 30, 2015, Mr. Osborne held outstanding options to purchase an aggregate of 74,490 shares of the Company's common stock at a weighted average exercise price of $9.53 per share, including unvested options to purchase 50,814 shares at a weighted average exercise price of $9.49 per share. |
| |
• | As described in Note 15, the Company designed a compensatory plan for its non-executive employees in connection with the relocation of its operations to Jersey City, New Jersey (the "Retention Plan"). Pursuant to the terms of the Retention Plan, all stock options held by non-executive employees eligible under the Retention Plan were modified to provide: (i) accelerated vesting of all unvested stock options as of December 31, 2015, and (ii) an extension to the existing 90-day post-employment option exercise period, which varies for each employee based upon years of service, with a maximum exercise period of 48 months. As of December 31, 2015, the retained employees eligible for participation in the Retention Plan held outstanding options to purchase 96,014 shares of the Company's common stock at a weighted average exercise price of $9.31 per share, including aggregate unvested options to purchase 64,279 shares at a weighted average exercise price of $9.20 per share. |
| |
• | In July 2015, pursuant to the Service Business Plan described in Note 15, the stock options held by each non-executive employee of the Services Business were modified immediately prior to the closing of the sale transaction in July 2015 to provide: (i) accelerated vesting of all unvested stock options as of the closing of the sale transaction and (ii) an extension to the existing 90-day post-employment option exercise period, which varies for each employee based upon years of service, with a maximum exercise period of 48 months. As of July 16, 2015, the non-executive employees of the Services Business held outstanding options to purchase 37,517 shares of the Company's common stock at a weighted average exercise price of $9.62 per share, including aggregate unvested options to purchase 23,052 shares at a weighted average exercise price of $9.61 per share. |
| |
• | On July 21, 2015, Yves J. Ribeill, Ph.D., President and a member of the Company’s board of directors, resigned as President. The Company and Dr. Ribeill entered into a Separation Agreement which included the following modifications to Dr. Ribeill's outstanding options to purchase the Company's common stock: (i) accelerated vesting of all unvested stock options as of July 21, 2015, and (ii) an extension to the existing 90-day post-employment option exercise period to 48 months. As of July 23, 2015, Dr. Ribeill held 84,613 vested options and 183,268 unvested options to purchase shares of the Company’s common stock at a weighted average exercise price of $9.61 and $9.41 per share, respectively. |
| |
• | On September 24, 2015, Edward E. Penhoet, Ph.D. resigned from the Company's board of directors. The Company's board of directors approved the following modifications to Dr. Penhoet's outstanding options to purchase the Company's common stock: (i) accelerated vesting of all unvested stock options as of September 24, 2015, and (ii) an extension to the existing 90-day post-employment option exercise period to 36 months. As of September 24, 2015, Dr. Penhoet held outstanding options to purchase 12,280 shares of the Company's common stock at a weighted average exercise price of $8.64 per share, including aggregate unvested options to purchase 8,800 shares at a weighted average exercise price of $8.65 per share. |
The Company determined the additional compensation cost associated with the previously described modifications pursuant to applicable guidance in FASB ASC Topic 718, Compensation—Stock Compensation. The additional compensation cost was determined by calculating the difference between (a) the estimated fair value of each option award immediately prior to the modifications and (b) the estimated fair value of each option award immediately after the modifications. The fair value of each option award immediately prior to and immediately after modification was estimated using the Black-Scholes option-pricing model to determine an incremental fair value, consistent with and in accordance with the Company’s existing accounting policy for stock compensation (see Note 2). Using the Black-Scholes option-pricing model, the weighted-average incremental fair value of outstanding modified option awards was $3.77 per option share. The total additional compensation cost associated with the previously described modifications was determined to be $1,869, which was expensed in the year ended December 31, 2015. The remaining additional compensation cost is associated with future service periods and will be recognized as those services are performed.
2014 Option Amendments
During the year ended December 31, 2014, the Company's board of directors approved the following with respect to the 2009 Stock Option Plan:
| |
• | On April 29, 2014, the exercise price per share of certain options to purchase 53,404 shares of common stock under the 2009 Stock Option Plan was lowered to an amount equal to $10.00 per share. The original exercise prices of such options ranged from $20.40 to $61.20 per share, with a weighted average exercise price of $54.87 per share. |
| |
• | On June 18, 2014, the exercise price per share of all outstanding options to purchase shares of common stock under the 2009 Stock Option Plan was lowered to an amount equal to $9.64 per share, the closing stock price on June 18, 2014. This modification lowered the exercise price of outstanding options to purchase 110,346 shares of common stock, including those options to purchase common stock that were previously modified on April 29, 2014. These outstanding stock options had exercise prices that ranged from $20.40 to $61.20 per share, with a weighted average exercise price of $41.87 per share. |
| |
• | Also on June 18, 2014, the contractual term of all outstanding options to purchase shares of common stock under the 2009 Stock Option Plan was extended to June 17, 2024. |
The Company determined the additional compensation cost associated with the previously described modifications pursuant to applicable guidance in FASB ASC Topic 718, Compensation—Stock Compensation. The additional compensation cost was determined by calculating the difference between (a) the estimated fair value of each option award immediately prior to the modifications and (b) the estimated fair value of each option award immediately after the modifications. The fair value of each option award immediately prior to and immediately after modification was estimated using the Black-Scholes option-pricing model, consistent with and in accordance with the Company’s existing accounting policy for stock compensation. Using the Black-Scholes option-pricing model, the weighted-average fair value of outstanding 2009 Stock Option Plan option awards was $3.08 per option immediately prior to modification on June 18, 2014 and was $5.87 per option immediately after modification. The additional compensation cost was determined to be $293, of which $130 was associated with services previously performed and, therefore, was expensed in the quarter ended June 30, 2014. The remaining additional compensation cost is being recognized as remaining services are performed.
Also on June 18, 2014, the board of directors approved modifications to the exercise price and contractual term of all outstanding option awards under the Company’s Stock Option Plan previously adopted by the Company in 1999 (the “1999 Stock Option Plan”). The modifications to the exercise price and contractual term are consistent with those previously described for outstanding options under the 2009 Stock Option Plan. In addition, the 1999 Stock Option Plan option awards were modified to provide that the holder may exercise vested shares under the option for the contractual term of the option even in the event the holder terminates services with the Company other than for cause. The modifications lowered the exercise price of outstanding options to purchase 73,087 shares of common stock, which had exercise prices that ranged from $20.40 to $25.50 per share, with a weighted average exercise price of $21.50 per share.
Pursuant to the terms of the 1999 Stock Option Plan, any amendments that modify the terms of the options awards require approval or consent of the Company’s shareholders. No additional compensation cost associated with the options under the 1999 Stock Option Plan was recognized during the quarter ended June 30, 2014 because the amendments were subject to and contingent upon stockholder approval. The Company did not believe stockholder approval was perfunctory. The Company’s stockholders approved the 1999 Stock Option Plan modifications on September 11, 2014, which was considered to be the measurement date for the determination of additional stock compensation expense. Consistent with the accounting guidance and methodology previously described for the 2009 Plan amendment, the Company determined the additional compensation cost associated with the 1999 Stock Option Plan modifications pursuant to FASB ASC Topic 718. The weighted-average fair value of outstanding 1999 Stock Option Plan option awards was $0.78 per option immediately prior to modification on September 11, 2014 and was $3.78 per option immediately after modification. The additional compensation cost was determined to be $225, all of which was associated with services previously performed and, therefore, was fully expensed in the quarter ended September 30, 2014.
2014 Employee Stock Purchase Plan
In February 2014, the Company’s board of directors adopted the 2014 Employee Stock Purchase Plan (“ESPP”), which was subsequently ratified by the Company’s stockholders and became effective on May 2, 2014. The purpose of the ESPP is to provide means by which eligible employees of the Company and of certain designated related corporations may be given an opportunity to purchase shares of the Company’s common stock, and to seek and retain services of new and existing employees and to provide incentives for such persons to exert maximum efforts for the success of the Company. Common stock that may be issued under the ESPP will not exceed 47,794 shares, plus the number of shares of common stock that are automatically added on January 1st of each year for a period of ten years, commencing on January 1, 2015 and ending on January 1, 2024, in an amount equal to the lesser of (i) 0.8% of the total number of shares of outstanding common stock on December 31 of the preceding calendar year, and (ii) 29,411 shares of common stock. Similar to the 2014 Plan, the board of directors may act prior to January 1st of a given year to provide that there will be no increase in the share reserve or that the increase will be a lesser number of shares than would otherwise occur. The ESPP is intended to qualify as an “employee stock purchase plan” within the meaning of Section 423 of the Internal Revenue Code.
During the year ended December 31, 2014, the Company issued 10,048 shares of common stock under the ESPP. During the year ended December 31, 2015, the number of shares of common stock available for issuance under the ESPP was automatically increased by 29,411 shares and the Company issued a total of 16,874 shares of common stock under the ESPP. As of December 31, 2015, there were 50,283 shares of common stock available for future issuance under the ESPP.
Compensation Cost
The compensation cost that has been charged against income for stock awards under the 2009 Stock Option Plan, the 2014 Plan, and the ESPP was $3,023 and $1,201 for the years ended December 31, 2015 and 2014, respectively. The total income tax benefit recognized in the statements of operations for share-based compensation arrangements was $0 for both the years ended December 31, 2015 and 2014. Cash received from options exercised was $0 and $9 for the years ended December 31, 2015 and 2014, respectively.
Stock-based compensation expense related to stock options is included in the following line items in the accompanying statements of operations:
|
| | | | | | | | |
| | Years Ended December 31, |
| | 2015 | | 2014 |
Research and development | | $ | 300 |
| | $ | 394 |
|
Selling, general and administrative | | 2,515 |
| | 648 |
|
Discontinued operations | | 208 |
| | 159 |
|
Total stock-based compensation expense | | $ | 3,023 |
| | $ | 1,201 |
|
The Company’s financial statements include total tax benefit of $0 and $1,166 on net losses from continuing operations before taxes of $28,338 and $6,769 for the years ended December 31, 2015 and 2014, respectively. Reconciliations of the differences between the benefit for income taxes and income taxes at the statutory U.S. federal income tax rate is as follows:
|
| | | | | | | | | | | | | |
| Years Ended December 31, |
| 2015 | | 2014 |
| Amount | | Percent of Pretax Income | | Amount | | Percent of Pretax Income |
Income taxes from continuing operations at statutory rate | $ | (9,635 | ) | | 34.0 | % | | $ | (2,301 | ) | | 34.0 | % |
State income taxes | (196 | ) | | 0.7 | % | | (471 | ) | | 7.0 | % |
Stock warrant derivative liability | — |
| | — | % | | (3,427 | ) | | 50.6 | % |
Stock-based compensation | 15 |
| | (0.1 | )% | | 268 |
| | (4.0 | )% |
R&D tax credits | (1,152 | ) | | 4.1 | % | | (320 | ) | | 4.7 | % |
Loss on sale of discontinued operations, net of reduction in related valuation allowances | (1,458 | ) | | 5.1 | % | | — |
| | — | % |
Other | (127 | ) | | 0.5 | % | | (190 | ) | | 2.8 | % |
Increase (decrease) in valuation allowance | 12,553 |
| | (44.3 | )% | | 5,275 |
| | (77.9 | )% |
Total income tax benefit | $ | — |
| | — | % | | $ | (1,166 | ) | | 17.2 | % |
The components of deferred tax assets and liabilities as of December 31, 2015 and 2014 are as follows:
|
| | | | | | | |
| December 31, |
| 2015 | | 2014 |
Current deferred tax assets: | | | |
Accrued expenses | $ | 783 |
| | $ | 1,052 |
|
Stock-based compensation | 1,507 |
| | 336 |
|
Assets of discontinued operations, net of liabilities | — |
| | 1,882 |
|
Other | 19 |
| | 15 |
|
| 2,309 |
| | 3,285 |
|
Noncurrent deferred tax assets (liabilities); | | | |
Net operating loss carryforwards | 42,358 |
| | 29,981 |
|
Research and development credits | 3,836 |
| | 2,684 |
|
| 46,194 |
| | 32,665 |
|
Total deferred tax assets | 48,503 |
| | 35,950 |
|
Valuation allowances | (48,503 | ) | | (35,950 | ) |
Net deferred tax assets | $ | — |
| | $ | — |
|
As of December 31, 2015 and 2014, the Company had available federal net operating loss ("NOL") carryforwards of approximately $117,272 and $81,687, respectively, North Carolina net economic loss ("NEL") carryforwards of approximately $117,721 and $85,790, respectively, and Pennsylvania NOL carryforwards of approximately $0 and $0, respectively. The federal NOL and North Carolina NEL carryforwards begin to expire in 2020 and 2015, respectively. As of December 31, 2015, the Company had available federal research and development credit carryforwards of $3,573 and North Carolina credit carryforwards of $255, which begin to expire in 2020 and 2015, respectively.
As of December 31, 2015 and 2014, the Company has concluded that it is more likely than not that the Company will not realize the benefit of its deferred tax assets due to its history of losses. Accordingly, the net deferred tax assets have been fully reserved.
In accordance with Section 382 of the Internal Revenue Code of 1986, as amended, a change in equity ownership of greater than 50% within a three-year period results in an annual limitation on the Company’s ability to utilize its NOL
carryforwards created during the tax periods prior to the change in ownership. The Company has determined that ownership changes have occurred and as a result, a portion of the Company’s NOL carryforwards are limited.
Because the Company has incurred cumulative net operating losses since inception, all tax years remain open to examination by U.S. federal and state income tax authorities.
The Company adopted FASB Accounting Standards Codification 740-10-25-5, Income Taxes, formerly FASB Interpretation No. 48, Accounting for Uncertainty in Income Taxes, as amended, on January 1, 2009. The difference between the tax benefit recognized in the financial statements and the tax benefit claimed in the tax return is referred to as an unrecognized tax benefit.
The following is a tabular reconciliation of the total amounts of unrecognized tax benefits as of December 31, 2015 and 2014:
|
| | | | | | | |
| December 31, |
| 2015 |
| 2014 |
Unrecognized tax benefit—January 1 | $ | 623 |
| | $ | 623 |
|
Additions for tax positions of current period | — |
| | — |
|
Additions for tax positions of prior periods | — |
| | — |
|
Other | — |
| | — |
|
Unrecognized tax benefit—December 31 | $ | 623 |
| | $ | 623 |
|
None of the unrecognized tax benefits would, if recognized, affect the effective tax rate because the Company has recorded a valuation allowance to fully offset federal and state deferred tax assets. The Company has no tax positions for which it is reasonably possible that the total amount of unrecognized tax benefits will significantly increase or decrease within the coming year. The Company has $0 provided for interest and penalties associated with uncertain tax positions.
The following table summarizes the computation of basic and diluted net loss per share attributable to the Company’s common stockholders:
|
| | | | | | | |
| Years Ended
December 31, |
| 2015 | | 2014 |
Income (loss) attributable to common stock - basic: | | | |
Loss from continuing operations | $ | (28,338 | ) | | $ | (5,603 | ) |
Deemed dividend for beneficial conversion feature on Series D-2 Preferred | — |
| | (909 | ) |
Deemed dividend for antidilution adjustments to convertible preferred stock | — |
| | (214 | ) |
Accretion of convertible preferred stock | — |
| | (510 | ) |
Loss from continuing operations attributable to common stock - basic | (28,338 | ) | | (7,236 | ) |
Income from discontinued operations, net of income tax expense, attributable to common stock - basic | (4,285 | ) | | 1,369 |
|
Net loss attributable to common stock - basic | $ | (32,623 | ) | | $ | (5,867 | ) |
| | | |
Income (loss) attributable to common stock - diluted: | | | |
Loss from continuing operations attributable to common stock - basic | $ | (28,338 | ) | | $ | (7,236 | ) |
Derivative fair value adjustment | — |
| | (10,080 | ) |
Loss from continuing operations attributable to common stock - diluted | (28,338 | ) | | (17,316 | ) |
Income from discontinued operations, net of income tax expense, attributable to common stock - diluted | (4,285 | ) | | 1,369 |
|
Net loss attributable to common stock - diluted | $ | (32,623 | ) | | $ | (15,947 | ) |
| | | |
Weighted-average common shares outstanding: | | | |
Weighted-average common shares outstanding - basic | 12,163,559 |
| | 5,663,311 |
|
Allocation of common stock warrants as participating securities | — |
| | 273,776 |
|
Weighted-average common shares outstanding - diluted | 12,163,559 |
| | 5,937,087 |
|
| | | |
Income (loss) per share - basic: | | | |
Continuing operations | $ | (2.33 | ) | | $ | (1.28 | ) |
Discontinued operations | $ | (0.35 | ) | | $ | 0.24 |
|
Net loss per share - basic | $ | (2.68 | ) | | $ | (1.04 | ) |
Income (loss) per share - diluted: | | | |
Continuing operations | $ | (2.33 | ) | | $ | (2.92 | ) |
Discontinued operations | $ | (0.35 | ) | | $ | 0.23 |
|
Net loss per share - diluted | $ | (2.68 | ) | | $ | (2.69 | ) |
The following securities, presented on a common stock equivalent basis, have been excluded from the calculation of weighted average common shares outstanding because their effect is anti-dilutive. As discussed in Note 8, in May 2014, upon completion of the IPO, all outstanding shares of the convertible preferred stock were converted into shares of common stock at their conversion prices. Therefore, as of December 31, 2015, the convertible preferred stock securities were no longer outstanding and will have no impact on net income or net loss per share. |
| | | | | |
| Years Ended
December 31, |
| 2015 | | 2014 |
Convertible preferred stock: | | | |
Series A Preferred | — |
| | 6,149 |
|
Series B Preferred | — |
| | 131,685 |
|
Series C Preferred | — |
| | 783,515 |
|
Series C-2 Preferred | — |
| | 173,213 |
|
Series D-1 Preferred | — |
| | 296,773 |
|
Series D-2 Preferred | — |
| | 300,549 |
|
Warrants to purchase Series C-1 Preferred | 14,033 |
| | 14,033 |
|
Stock options | 1,379,727 |
| | 615,322 |
|
ESPP | — |
| | 65,401 |
|
| |
13. | Related-Party Transactions |
The Company had transactions with related parties as follows:
|
| | | | | | | |
| Years Ended December 31, |
| 2015 | | 2014 |
Revenue | $ | 2,140 |
| | $ | 7,288 |
|
Selling, general and administrative expense | $ | — |
| | $ | 500 |
|
Research Services Agreement with a Related-Party
Sanofi owns 100% of a subsidiary, Merial, which was a customer of the Services Business, presented in discontinued operations in the accompanying statements of operations (see Note 18). Both Sanofi and the subsidiary have an investment in the Company. The Company’s related-party revenue with Merial composed 29% and 41% of total revenue in discontinued operations for the years ended December 31, 2015 and 2014, respectively.
Success Fee Paid to a Related Party
In May 2014, the Company paid a $500 success fee to Burrill Securities, an affiliate of Burrill Biotechnology Capital Fund, L.P., a holder of the Company’s capital stock, pursuant to an engagement letter. The fee was recognized as general and administrative expense in the accompanying statements of operations.
14. Employee Benefit Plan
The Company has a 401(k) retirement plan, which covers all U.S. employees scheduled for and working more than 20 hours per week. The Company may provide a discretionary match with a maximum amount of 50% of the first 6% of eligible participant’s compensation, which vests ratably over four years. Contributions under the plan during were approximately $183 and $239 during the years ended December 31, 2015 and 2014, respectively, including $104 and $176 presented in discontinued operations in the accompanying statements of operations for the years ended December 31, 2015 and 2014, respectively.
15. Compensatory Plan Obligations
Compensatory Plan with Services Business Employees
In connection with the Company's sale of its Services Business, which is more fully described in Note 18, the Company designed a compensatory plan to promote the retention of services of its non-executive employees supporting that business (the "Services Business Plan"). The Company's board of directors adopted, and the Company communicated, the material terms of the Services Business Plan prior to June 30, 2015, to all non-executive employees of the Services Business. The Services Business Plan terms provided for certain cash compensation payments, as well as modifications to the terms of currently outstanding stock options held by such non-executive employees, as more completely described below, upon the successful closing of the sale of the Services Business. The sale closed in July 2015 (see Note 18). The Services Business Plan meets the definition of an exit and disposal activity pursuant to FASB ASC 420--Exit and Disposal Cost Obligations and all related expenses incurred have been presented in discontinued operations in the statements of operations in the period incurred.
The Services Business Plan provided that in the event a non-executive employee of the Services Business was not offered a comparable position by Accuratus, the Company would provide severance payments to such employees. The Company terminated certain employees in June 2015 (the "June 2015 Terminated Employees") who became eligible for severance benefits totaling approximately $999 pursuant to the terms of the Services Business Plan, which was expensed in the quarterly period ended June 30, 2015. As of December 31, 2015, the remaining severance obligation for the June 2015 Terminated Employees was $319, which was included in accrued severance and retention liabilities in the accompanying balance sheet. The Services Business Plan also provided for certain amendments to the terms of the outstanding stock option awards held by the June 2015 terminated employees, which are described in Note 10.
In July 2015, pursuant to the Services Business Plan, the Company paid cash totaling approximately $215 to certain non-executive employees of the Services Business representing an incentive payment upon the closing of the sale of the Services Business. In addition, all non-executive employees of the Services Business were eligible to receive a cash retention compensation payment from the Company on the earlier of (i) the six month anniversary of the closing of the sale transaction, provided that they remained employed by Accuratus as of such date, or (ii) the date of termination of such employee by Accuratus without good cause. Maximum cash retention compensation payments could have totaled approximately $814 under the Services Business Plan, if all service business employees had remained eligible pursuant to the terms of the Services Business Plan. The Company incurred these obligations on the date of the sale of the Services Business in July 2015; therefore, the estimated fair value of the compensation expense associated with these cash payments and obligations was recognized during the quarterly period ended September 30, 2015. As of December 31, 2015, after adjusting for employees who forfeited their benefits under the Services Business Plan, the Company recorded a liability of $763, which was included in accrued severance and retention liabilities in the accompanying balance sheet. This liability was satisfied in January 2016 when the Company paid all cash retention compensation payments to the former employees.
The Services Business Plan also includes certain amendments to the terms of the eligible employees' outstanding stock option awards, which are described in Note 10.
Compensatory Arrangement with Employees of the Company's Continuing Operations
In connection with the Company's relocation of its continuing operations to Jersey City, New Jersey, the Company designed a compensatory plan to promote the retention of services of non-executive employees supporting its continuing operations (the "Retention Plan"). The Company's board of directors adopted, and the Company communicated, the material terms of the Retention Plan prior to June 30, 2015, to all non-executive employees supporting the Company's continuing operations. The Retention Plan terms provided for certain cash compensation payments and severance payments, as well as modifications to the terms of currently outstanding stock options held by such non-executive employees, as more completely described below. The Company has concluded that the Retention Plan meets the definition of an exit and disposal activity pursuant to FASB ASC 420--Exit and Disposal Cost Obligations as of June 30, 2015, and all related expenses incurred have been presented in continuing operations in the statements of operations.
The Retention Plan provided that non-executive employees were eligible to receive cash bonuses, severance payments and related benefit premiums, provided that all current employees remained employed through December 31, 2015 and were not terminated for cause. The Retention Plan also provided that if the Company and an employee agreed upon a services termination date earlier than December 31, 2015 (the "Release Date"), the employee would remain eligible for all terms of the Retention Plan. The Company accrued this obligation over the remaining future service period required by the employees through the earlier of the Release Date or December 31, 2015. During the year ended December 31, 2015, the Company recognized total expense of $1,012, which was included in research and development and selling, general, and administrative expenses in the accompanying statements of operations. The corresponding liability is included in accrued severance and retention obligations in the accompanying balance sheet.
The Retention Plan also includes certain amendments to the terms of the eligible employees' outstanding stock option awards, which are described in Note 10.
Compensatory Arrangements with Former Executive Officers
Charles F. Osborne, Jr., the Company’s former chief financial officer, resigned from the Company effective June 30, 2015. The Company's compensation committee of the board of directors approved a compensatory arrangement for Mr. Osborne that provided for certain payments and benefits, including: (i) a cash payment of approximately $138 upon his resignation on June 30, 2015; (ii) cash severance payments totaling approximately $179, which was equal to seven months of Mr. Osborne’s then effective base salary, paid over seven months commencing with the first payroll period following the resignation date; (iii) a payment representing a contribution Mr. Osborne can use towards continuing COBRA premiums for medical, dental, and vision group health coverage for a period up to seven months after the resignation date; and (iv) certain amendments to the terms of Mr. Osborne's outstanding stock option awards (see Note 10). The cash severance payments and related benefit premiums and payroll taxes totaled approximately $335 and were expensed in the quarterly period ended June 30, 2015. As of December 31, 2015, the remaining obligation of $26 is included in accrued severance and retention liabilities in the accompanying balance sheet.
Yves J. Ribeill, Ph.D., President and a member of the Company’s board of directors, resigned as President effective July 21, 2015. Dr. Ribeill continues to serve on the board of directors. The Company and Dr. Ribeill entered into an agreement, effective July 21, 2015, (the “Separation Agreement”), providing for certain payments and benefits to Dr. Ribeill, including: (i) a cash payment of approximately $100 upon the effective date of his resignation; (ii) cash severance payments totaling approximately $900, paid over 12 months commencing with the first payroll period following the resignation date; (iii) a payment representing a contribution Dr. Ribeill can use towards continuing COBRA premiums for medical, dental, and vision group health coverage after the resignation date; and (iv) certain amendments to the terms of Dr. Ribeill's outstanding stock option awards (see Note 10). The cash severance payments and related benefit premiums and payroll taxes totaled approximately $1,046 as of July 21, 2015, which was recognized as expense in the quarterly period ended September 30, 2015. As of December 31, 2015, the remaining obligation of $502 is included in accrued severance and retention liabilities in the accompanying balance sheet.
| |
16. | Fair Value Measurements |
The carrying amounts of certain financial instruments, including cash and cash equivalents, accounts receivable, unbilled services, prepaid expenses and other current assets, accounts payable, and accrued expenses approximate their respective fair values due to the short-term nature of such instruments.
Assets and Liabilities Measured at Fair Value on a Non-Recurring Basis
As discussed in Note 18, the Company met the relevant criteria for reporting the Service Business as held for sale on May 4, 2015 (the "Measurement Date"), and as a result, assessed the asset group for impairment pursuant to FASB Topic 360, Property, Plant, and Equipment. The net carrying value of the Services Business asset group was compared to its fair value as of May 4, 2015. The Company determined that the selling price paid by Accuratus to acquire the Services Business asset group was the best estimate of fair value, which the Company concluded was a Level 2 input. The Company determined that the Services Business asset group's net carrying value exceeded its fair value by $572 on the Measurement Date. The Company also estimated selling costs directly attributable to the sale of the Services Business to be $778. As a result, the Company recorded a $1,350 impairment charge on property and equipment assets classified as held for sale in the quarterly period ended June 30, 2015. The Company subsequently recorded a $73 loss on disposal, after the effects of $764 of actual selling costs, in the quarterly period ended September 30, 2015, due to (i) a difference between estimated and final direct selling costs and (ii) a change in estimated working capital of the Services Business between June 30, 2015 and the effective date of the sale on July 17, 2015.
Assets and Liabilities Measured at Fair Value on a Recurring Basis
The Company evaluates its financial assets and liabilities subject to fair value measurements on a recurring basis to determine the appropriate level in which to classify them for each reporting period, pursuant to the policy described in Note 2. This determination requires significant judgments to be made.
The following table summarizes the conclusions reached as of December 31, 2015 and 2014 for financial instruments measured at fair value on a recurring basis:
|
| | | | | | | | | | | | |
| | | | Fair Value Hierarchy Classification |
| | Balance | | Quoted Prices in Active Markets for Identical Assets (Level 1) | | Significant Other Observable Inputs (Level 2) | | Significant unobservable inputs (Level 3) |
December 31, 2014 | | | | | | | | |
Cash on deposit | | 32,243 |
| | 32,243 |
| | — |
| | — |
|
Money market funds | | — |
| | — |
| | — |
| | — |
|
Total cash and cash equivalents | | 32,243 |
| | 32,243 |
| | — |
| | — |
|
| | | | | | | | |
December 31, 2015 | | | | | | | | |
Cash on deposit | | 46,935 |
| | 46,935 |
| | — |
| | — |
|
Money market funds | | 50 |
| | 50 |
| | — |
| | — |
|
Total cash and cash equivalents | | 46,985 |
| | 46,985 |
| | — |
| | — |
|
The Company measures cash equivalents at fair value on a recurring basis. The fair value of cash equivalents is determined based on “Level 1” inputs, which consist of quoted prices in active markets for identical assets.
The Company’s derivative liabilities associated with the Series D-2 Preferred sale on January 31, 2014 were measured at fair value on a recurring basis. The fair value of these warrant derivatives was based on a valuation of the Company’s common stock. In order to determine the fair value of the Company’s common stock, the Company used a probability-weighted expected return method, or PWERM. Significant inputs for the PWERM included an estimate of the Company’s equity value, a weighted average cost of capital and an estimated probability and timing for each valuation scenario.
A reconciliation of the beginning and ending balances for liabilities measured at fair value on a recurring basis using significant unobservable inputs (Level 3) is as follows:
|
| | | |
| Year Ended |
| December 31, 2014 |
Balance - January 1, 2014 | $ | 12,237 |
|
Issuance of warrants | 544 |
|
Excess of fair value of warrants over proceeds | 362 |
|
Adjustment to fair value | (10,442 | ) |
Reclassification to additional paid-in capital upon exercise of warrants | (2,701 | ) |
Balance - December 31, 2014 | $ | — |
|
| |
17. | Significant Agreements |
R-Pharm Collaboration Arrangement
In August 2013, the Company entered into a development, license, and supply agreement (the “original agreement”) with R-Pharm, granting it exclusive rights to develop and commercialize SCY-078, the Company's lead antifungal compound, in the field of human health in Russia and certain smaller non-core markets. The Company received an upfront payment of $1,500, the unamortized portion of which comprises its deferred revenue balance as of December 31, 2015, and is entitled to receive payments on contingent events, including 1) a development milestone payment of $3,000 upon the first registration of SCY-078 in any country covered by the agreement; 2) sales-based payments of up to $15,000 upon R-Pharm’s achievement of specified targets for cumulative net sales of SCY-078; and 3) percentage royalties of up to the mid-teens on SCY-078 net sales.
The Company deferred the upfront payment received and is recognizing it over the estimated relationship period of 70 months, which includes the product development period and an additional period during which the Company is required to participate in a product development committee. The development milestone payment is considered substantive and will be recognized when R-Pharm achieves certain specified milestones.
The sales-based payments will not be recognized until the Company 1) receives the payments, and 2) has no continuing performance obligations. If the Company has any continuing performance obligations when the sales-based payments are received, those payments will be deferred and recognized over the remaining period of continuing performance obligations. Royalties will be recognized when payment is received.
The original agreement also included terms whereby R-Pharm would reimburse the Company for certain research and development costs associated with Phase 2 and Phase 3 clinical trials of oral SCY-078 and the development of an IV formulation of SCY-078. However, these cost reimbursement terms required that the clinical trials and the IV formulation development follow a global development plan that was agreed upon by both parties in August 2013. Subsequent to August 2013, modifications were made to the global development plan that caused the clinical trial cost reimbursement terms in the original agreement to no longer be enforceable. As a result, the Company concluded that persuasive evidence of a cost reimbursement arrangement did not exist under the original agreement with R-Pharm. Further, the IV formulation development cost reimbursement terms in the original agreement did not specify which IV formulation and development costs were reimbursable by R-Pharm. Because of this lack of specificity, the Company concluded that the reimbursable fees due from R-Pharm were not determinable under the original agreement.
In November 2014, the Company entered into a supplemental arrangement with R-Pharm, whereby R-Pharm was informed of the modified IV formulation development plan and R-Pharm agreed to reimburse the Company for specifically identified IV formulation development and manufacturing costs incurred by the Company. The specifically identified costs were defined as all costs incurred by the Company under a separate arrangement between the Company and a third-party service provider, whereby the third-party service provider is performing certain IV formulation and development services. The Company concluded that the original agreement, when combined with the November 2014 supplemental arrangement, provided persuasive evidence of a cost reimbursement arrangement between the Company and R-Pharm as of December 31, 2014. Therefore, the Company has recognized receivables due from R-Pharm and has received reimbursement payments from R-Pharm during 2014 and 2015. The presentation and disclosure associated with this cost reimbursement receivable is in accordance with the Company's research and development expenses accounting policy described in Note 2.
The agreement with R-Pharm relates to the Company's continuing operations and was not associated with the sale of the Services Business in July 2015 (see Note 18).
Elanco Licensing Agreement
The Company entered into a licensing agreement with Elanco Animal Health (Elanco) in December 2013. The Elanco Licensing Agreement is directly related to the Services Business and was assigned to Accuratus in conjunction with the sale of the Services Business in July 2015 (see Note 18), including all future obligations and benefits as summarized below. Revenue from the agreement is included in discontinued operations in the accompanying statements of operations.
The agreement included an upfront payment of $500 and multi-year contract research and development services with fees of $2,750 annually for the first two years and $3,000 annually for the second two years, and entitles the Company to 1) development milestone payments of up to $1,500 for each compound Elanco and the Company decided to develop; 2) a one-time payment of up to $2,000 for the first regulatory approval of any product in the U.S.; 3) a one-time payment of $4,000 for the first commercial sale of a product in the U.S. and a one-time payment of $1,500 for the first commercial sale of a product in the European Union; 4) one-time payments of up to $15,000 for reaching specified annual sales of a product; and 5) mid-single-digit percentage royalties on net annual sales. The Company deferred the upfront payment, which it received in January 2014, and was recognizing the revenue over the research and development period of four years.
Waterstone Licensing Agreement
On October 29, 2014, the Company entered into a license agreement with Waterstone Pharmaceutical (HK Limited), or Waterstone, under which the Company granted Waterstone an exclusive, worldwide license to develop and commercialize SCY-635 for the treatment of viral diseases in humans. In addition, under the same agreement, the Company granted Waterstone an option for an exclusive, worldwide license to develop and commercialize two additional compounds of the Company, SCY-575 and SCY-116, for the treatment of viral diseases in humans. The option is exercisable for a period of 18 months from the date of the agreement. In addition, the Company agreed that during the term of the agreement, it would not develop or commercialize, or grant any right or license to any third party to develop or commercialize, in Asia (excluding Japan), any cyclophilin inhibitor for treatment of viral diseases in humans.
The agreement expires upon Waterstone’s last royalty payment, which is the later of ten years from the last registration of the product, or the last to expire of the patents. Either party may terminate the agreement if the other party breaches and fails to remedy the breach after receiving notice from the nonbreaching party. Specifically, the Company has the ability to terminate the agreement if the Company determines that Waterstone failed to make reasonable progress in the development and commercialization of SCY-635 or the optioned compounds. If the Company gives Waterstone notice of failure to make
reasonable progress, Waterstone will have the opportunity to correct the deficiencies. If Waterstone fails to do so, the Company has the right to terminate the license.
The Company received a non-refundable upfront license fee payment of $1,000 in November 2014 for SCY-635, and may receive an additional upfront payment of $500 if Waterstone exercises its option for the two additional compounds. The Company is also entitled to receive certain payments on contingent future events, including 1) a development milestone payment of $4,000 upon the first registration of a product, and 2) royalties based on a specified percentage of net sales (which percentage is in the mid-single digits), varying based on whether the product contains SCY-635 or one of the two additional compounds.
The Company analyzed the license agreement and concluded that, as of December 31, 2014, it had no remaining substantive obligations to perform under the arrangement. As a result, the Company recognized revenue of $1,000 from the non-refundable upfront payment in the year ended December 31, 2014. The development milestone payment and the royalties will be recognized as revenue if and when the Company receives the payments.
The Waterstone Licensing Agreement relates to the Company's continuing operations and was not associated with the sale of the Services Business in July 2015 (see Note 18).
Merial Research Services Agreement
In December 2014, the Company entered into an agreement with Merial, a related party (see Note 13), under which the Company provided contract research and screening services in the field of animal health that primarily target parasites. The Merial Research Services Agreement is directly related to the Services Business and was assigned to Accuratus in conjunction with the sale of the Services Business in July 2015 (see Note 18), including all future obligations and benefits as summarized below. Revenue from the agreement is included in discontinued operations in the accompanying statements of operations.
Prior to the execution of this agreement, the Company provided contract research and development services for Merial on a fee-for-service basis under a separate agreement that expired on December 31, 2014. The agreement was a non-exclusive arrangement in the animal health field and is on a fee-for-service basis. The Company could not receive any contingent payments based on the progression to development and commercialization of any compounds arising from this agreement. Any intellectual property created in connection the Company's performance of the services would be the sole property of Merial. The term of the agreement was two years, beginning January 1, 2015 and ending on December 31, 2016, and the total service fee due from Merial over the term of the agreement was $7,900, payable in equal quarterly installments. The agreement also provided for an option to extend the term for one additional year.
Either party could terminate the agreement in the event of breach of material obligation by the other party if such breach is not remedied after written notice from the non-breaching party. Either party could terminate this agreement if the other party makes an assignment for the benefit of creditors, becomes subject to bankruptcy proceedings, subject to appointment of a receiver, or admits inability to pay its debts. Further, within six months of any change of control the Company, Merial shall either (i) consent to continue the agreement pursuant to its terms, (ii) agree to an assignment of the agreement to a third-party acceptable to Merial, or (iii) the parties shall implement another solution acceptable to Merial, provided, however, if no resolution acceptable to Merial has been implemented within six months of the change of control, Merial may terminate the agreement immediately. If Merial believes in good faith that the Company acted in any way that may subject Merial to liability under anti-corruption laws, Merial had the unilateral right to terminate this agreement. At termination or expiration of the agreement for any reason, upon Merial’s request, the Company was required to transfer all agreement intellectual property to Merial.
| |
18. | Discontinued Operations |
On May 4, 2015, the Company's board of directors directed management to pursue a plan to sell the Service Business to Accuratus, representing a strategic shift in the Company's operations. The Company met the relevant criteria for reporting the service business as held for sale and in discontinued operations in the second quarter of 2015, pursuant to FASB Topic 205-20, Presentation of Financial Statements--Discontinued Operations, and FASB Topic 360, Property, Plant, and Equipment. The Company assessed the Services Business net asset group for impairment pursuant to FASB Topic 360 and recorded a $1,350 impairment charge on classification of property and equipment assets as held for sale in the quarterly period ended June 30, 2015. The fair value measurement used to determine the impairment charge has been described in Note 16.
Sale of the Services Business
On July 21, 2015, the Company completed the sale of the Services Business to Accuratus pursuant to the Purchase Agreement, with an effective date of July 17, 2015 for an aggregate purchase price of $3,875, subject to a working capital adjustment of $824, which reduced the proceeds at closing. In addition, a portion of the consideration payable at closing equal to $500 was withheld and is subject to an escrow for a period of 12 months from the date of closing to satisfy indemnification
obligations of the Company in connection with breaches of any representation and warranties and other customary obligations under the terms of the Purchase Agreement. The Company has not identified any breaches or other events that would cause a reduction in the escrow funds expected to be received by the Company. The escrow funds were recorded as a receivable included in prepaid expenses and other current assets in the accompanying balance sheets. The net cash consideration received by the Company upon closing in July 2015 was $2,549, after adjusting for the items described above and a nominal escrow fee.
The following table describes the net proceeds from the sale and the assets and liabilities sold, net of impairment charges and loss on disposal:
|
| | | | |
| | July 16, 2015 |
Net proceeds from sale of the Services Business | | |
Net cash consideration received at closing | | $ | 2,549 |
|
Consideration in escrow | | 500 |
|
Total consideration | | 3,049 |
|
Less: selling costs | | 764 |
|
Proceeds from sale, net of selling costs | | $ | 2,285 |
|
| | |
Services Business assets and liabilities disposed of on July 16, 2015 | | |
Accounts and unbilled receivables, net | | $ | 1,470 |
|
Prepaid expenses and other current assets | | 713 |
|
Property and equipment, net of accumulated depreciation | | 4,900 |
|
Other assets | | 59 |
|
Assets of Services Business, net | | $ | 7,142 |
|
| | |
Accounts payable and accrued expenses | | $ | 616 |
|
Deferred revenue | | 1,657 |
|
Deferred rent | | 1,161 |
|
Liabilities related to assets of the Services Business | | $ | 3,434 |
|
| | |
Assets of the Services Business, net of liabilities | | $ | 3,708 |
|
Less: Impairment charge recognized upon classification as held for sale | | 1,350 |
|
Less: Loss on disposal | | 73 |
|
Assets of the Services Business, net of liabilities and impairment charges | | $ | 2,285 |
|
Continuing Involvement with Accuratus
As a condition to the execution of the Purchase Agreement, Accuratus assumed the Company’s post-closing obligation under its facility lease in Durham, North Carolina. The Company and its retained employees continued to operate from the Durham facility immediately after the closing for a period of up to 6 months pursuant to a facility lease agreement. In addition, under a Transition Services Agreement, Accuratus provided accounting, IT, payroll, personnel and human resources support, and equity compensation plan administration support services to the Company at rates ranging from one hundred to two hundred dollars per hour for a period of time not to extend beyond December 31, 2015.
The Company and Accuratus also entered into the Services Agreement pursuant to which Accuratus will provide the Company with certain contract research and development services for 18 months (the "Initial Term") following the closing of the sale of the Services Business for a minimum purchase obligation of at least $3,300 due from the Company over the Initial Term of the Services Agreement. The purpose of the Services Agreement is to replace services that were previously provided internally by employees of the Company prior to the sale of the Services Business. The employees performing these services became employees of Accuratus in connection with this sale transaction.
During the year ended December 31, 2015, the Company recognized $1,602 of expense for services provided by Accuratus under the Services Agreement, which is included in research and development expense in the accompanying audited statements of operations.
Discontinued Operations and Assets Held for Sale
The following table presents a reconciliation of the carrying amounts of assets and liabilities of the Services Business to assets held for sale, net in the balance sheet:
|
| | | | |
| | December 31, 2014 |
Carrying amounts of assets included as part of discontinued operations: | | |
Accounts and unbilled receivables, net | | $ | 1,501 |
|
Prepaid expenses and other current assets | | 289 |
|
Property and equipment, net | | 4,835 |
|
Other assets | | 76 |
|
Assets of discontinued operations, net | | $ | 6,701 |
|
| | |
Carrying amounts of liabilities included as part of discontinued operations: | | |
Accounts payable and accrued expenses | | $ | 681 |
|
Deferred revenue | | 445 |
|
Deferred rent | | 1,294 |
|
Liabilities related to assets of discontinued operations | | $ | 2,420 |
|
The following table presents revenue, (expenses), gains, and (losses) attributable to discontinued operations:
|
| | | | | | | | |
| | Years Ended December 31, |
| | 2015 | | 2014 |
Major line items constituting income of discontinued operations: | | | | |
Total revenue | | $ | 7,408 |
| | $ | 17,768 |
|
Cost of revenue | | (7,296 | ) | | (15,446 | ) |
Research and development | | (860 | ) | | — |
|
Selling, general, and administrative | | — |
| | 48 |
|
Gain on insurance recovery | | — |
| | 165 |
|
Severance and exit costs (Note 15) | | (2,114 | ) | | — |
|
Impairment charge from classification of assets as held for sale | | (1,350 | ) | | — |
|
Gain (loss) on disposal, net of associated transaction costs of $764 | | (73 | ) | | — |
|
Income tax expense | | — |
| | (1,166 | ) |
Income (loss) from discontinued operations, net of income tax expense | | $ | (4,285 | ) | | $ | 1,369 |
|
The following table presents depreciation, capital expenditures, and significant operating and investing non-cash items related to the discontinued operations:
|
| | | | | | | |
| Years Ended December 31, |
| 2015 | | 2014 |
Depreciation expense | $ | 391 |
| | $ | 1,132 |
|
Purchases of property and equipment | (547 | ) | | (704 | ) |
Proceeds from insurance recovery | — |
| | 216 |
|
Gain on insurance recovery | — |
| | (165 | ) |
Stock-based compensation | 208 |
| | 159 |
|
Changes in deferred rent | (133 | ) | | (187 | ) |
Equipment purchases in accounts payable and accrued expenses | — |
| | 34 |
|
Impairment of fixed asset | — |
| | 51 |
|
Equity Incentive Plan Activities
Pursuant to the terms of the 2014 Plan (see Note 10), on January 1, 2016, the Company automatically added 556,223 shares to the total number shares of common stock available for future issuance under the 2014 Plan.
Pursuant to the terms of the 2014 ESPP (see Note 10), on January 1, 2016, the Company automatically added 29,411 shares to the total number shares of common stock available for future issuance under the 2014 ESPP.
| |
ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE |
None.
| |
ITEM 9A. | CONTROLS AND PROCEDURES |
Management’s Evaluation of our Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in the reports that we file or submit under the Securities Exchange Act of 1934 is (1) recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms and (2) accumulated and communicated to our management, including our principal executive officer and principal financial officer, to allow timely decisions regarding required disclosure.
As of December 31, 2015, our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934). Our management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives, and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Our principal executive officer and principal financial officer have concluded based upon the evaluation described above that, as of December 31, 2015, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Our internal control over financial reporting is designed to provide reasonable assurance to our management and board of directors regarding the preparation and fair presentation of published financial statements. A control system, no matter how well designed and operated, can only provide reasonable, not absolute, assurance that the objectives of the control system are met. Because of these inherent limitations, management does not expect that our internal controls over financial reporting will prevent all error and all fraud. Under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework in Internal Control—Integrated Framework (2013 Framework) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework in Internal Control—Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2015.
This annual report does not include an attestation report of our independent registered public accounting firm regarding our internal control over financial reporting due to an exemption provided by the JOBS Act for emerging growth companies.
Changes in Internal Control Over Financial Reporting
In connection with the relocation of our corporate offices from North Carolina to New Jersey, we hired new personnel within our accounting and finance department, including a new director of SEC reporting and a corporate controller. We began transitioning all accounting and finance processes and internal control activities to these new employees in December 2015 and the transition was substantially complete upon the filing of our 2015 Form 10-K in March 2016. During the transition process, while we continued to execute our existing system of internal controls over financial reporting, we made certain enhancements relating to our internal control over financial reporting as part of our compliance with internal control requirements of the Sarbanes-Oxley Act of 2002. Except for the previously described changes, during the quarter ended December 31, 2015, there have been no changes in our internal control over financial reporting, as such term is defined in Rules 13a-15(f) and 15(d)-15(f) promulgated under the Securities Exchange Act of 1934, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| |
ITEM 9B. | OTHER INFORMATION |
None.
PART III
| |
ITEM 10. | DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE |
The information required by this Item 10 is incorporated herein by reference from our Proxy Statement, which will be filed with the SEC within 120 days after the end of our 2015 fiscal year pursuant to Regulation 14A for our 2016 Annual Meeting of Stockholders (the “Proxy Statement”), under the captions “Executive Officers of the Company,” “Proposal 1 - Election of Directors,” “Section 16 Beneficial Ownership Reporting Compliance,” and “Code of Business Conduct and Ethics.”
A printed copy of the Proxy Statement will be sent, without charge, to any shareholder who requests it by writing to the Chief Financial Officer of Scynexis, Inc., 101 Hudson Street, Suite 3610, Jersey City, NJ 07302 - 6548.
| |
ITEM 11. | EXECUTIVE COMPENSATION |
The information required by this Item 11 is incorporated herein by reference from the Proxy Statement under the captions “Executive Compensation” and “Director Compensation.”
| |
ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS |
The information required by this Item 12 is incorporated herein by reference from the Proxy Statement, under the captions “Security Ownership of Certain Beneficial Owners and Management” and “Equity Compensation Plan Information.”
| |
ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE |
The information required by this Item 13 is incorporated herein by reference from the Proxy Statement, which will be filed with the SEC within 120 days after the end of the Company’s 2015 fiscal year pursuant to Regulation 14A for its 2016 Annual Meeting of Stockholders.
| |
ITEM 14. | PRINCIPAL ACCOUNTING FEES AND SERVICES |
The information required by this item will be set forth in our 2016 Proxy Statement under the caption “Principal Accountant Fees and Services.”
PART IV
| |
ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES |
Documents filed as part of this report:
| |
1. | List of Financial Statements |
The financial statements required by this item are listed in Item 8, “Financial Statements and Supplementary Data” herein.
| |
2. | List of Financial Statement Schedules |
All schedules are omitted because they are not applicable, not required or the required information is shown in the consolidated financial statements or notes thereto.
See the Exhibit Index which follows the signature page of this Annual Report on Form 10-K, which is incorporated herein by reference.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
| | |
SCYNEXIS, INC. |
| |
By: | | /s/ Marco Taglietti M.D. |
| | Marco Taglietti, M.D. |
| | Chief Executive Officer (Principal Executive Officer) |
| |
Date: | | March 7, 2016 |
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated. |
| | | | |
Signature | | Title | | Date |
| | | | |
/s/ Marco Taglietti M.D. | | Chief Executive Officer | | March 7, 2016 |
Marco Taglietti M.D. | | (Principal Executive Officer) | | |
| | | | |
/s/ Eric Francois | | Chief Financial Officer | | March 7, 2016 |
Eric Francois | | (Principal Financial and Accounting Officer) | | |
| | | | |
/s/ Guy Macdonald | | Director | | March 7, 2016 |
Guy Macdonald | | | | |
| | | | |
/s/ C. Patrick Machado | | Director | | March 7, 2016 |
C. Patrick Machado | | | | |
| | | | |
/s/ David Hastings | | Director | | March 7, 2016 |
David Hastings | | | | |
| | | | |
/s/ Steven C. Gilman, Ph.D. | | Director | | March 7, 2016 |
Steven C. Gilman, Ph.D. | | | | |
| | | | |
/s/ Ann F. Hanham, Ph.D. | | Director | | March 7, 2016 |
Ann F. Hanham, Ph.D. | | | | |
| | | | |
/s/ Patrick J. Langlois, Ph.D. | | Director | | March 7, 2016 |
Patrick J. Langlois, Ph.D. | | | | |
| | | | |
/s/ Yves J. Ribeill Ph.D. | | Director | | March 7, 2016 |
Yves J. Ribeill, Ph.D. | | | | |
INDEX TO EXHIBITS
|
| | |
Exhibit Number | | Description of Document |
| |
2.1 | | Asset Purchase Agreement, dated July 17, 2015, between SCYNEXIS, Inc. and Accuratus Lab Services, Inc. (Filed with the SEC as Exhibit 10.1 to our current report on Form 8-K, filed with the SEC on July 23, 2015, SEC File No. 001-36365). |
| |
3.1 | | Amended and Restated Certificate of Incorporation. (Filed with the SEC as Exhibit 3.1 to our Current Report on Form 8-K, filed with the SEC on May 12, 2014, SEC File No. 001-36365). |
| |
3.3 | | Amended and Restated Bylaws, as amended and as currently in effect. (Filed with the SEC as Exhibit 3.4 to our Registration Statement on Form S-1, filed with the SEC on February 27, 2014, SEC File No. 333-194192). |
| |
4.1 | | Reference is made to Exhibits 3.1 and 3.2 |
| |
4.2 | | Fifth Amended and Restated Investor Rights Agreement, dated December 11, 2013 (Filed with the SEC as Exhibit 10.21 to our Registration Statement on Form S-1, filed with the SEC on February 27, 2014, SEC File No. 333-194192). |
| |
10.1 | | Form of Indemnity Agreement between the Registrant and its directors and officers. (Filed with the SEC as Exhibit 10.1 to our Amendment No. 1 to Registration Statement on Form S-1, filed with the SEC on March 19, 2014, SEC File No. 333-194192). |
| |
10.2* | | SCYNEXIS, Inc. Stock Option Plan, as amended, and Forms of Stock Option Grant Notice, Stock Option Agreement and Notice of Stock Option Exercise. (Filed with the SEC as Annex B to our Proxy Statement on Schedule 14A, filed with the SEC on August 1, 2014, SEC File No. 001-36365). |
| | |
10.3* | | SCYNEXIS, Inc. 2009 Stock Option Plan, as amended, and Forms of Stock Option Grant Notice, Stock Option Agreement and Notice of Stock Option Exercise. (Filed with the SEC as Exhibit 10.3 to our Amendment No. 1 to Registration Statement on Form S-1, filed with the SEC on March 19, 2014, SEC File No. 333-194192). |
| |
10.4* | | SCYNEXIS, Inc. 2014 Equity Incentive Plan, as amended, (Filed with the SEC as Annex A to our proxy statement on Schedule 14A, filed with the SEC on April 22, 2015, SEC File No. 001-36365). |
| |
10.5* | | SCYNEXIS, Inc. 2014 Employee Stock Purchase Plan. (Filed with the SEC as Exhibit 99.4 to our Registration Statement on Form 8, filed with the SEC on May 16, 2014, SEC File No. 333-196007). |
| |
10.6* | | Compensation Arrangement with Non-Employee Directors. (Filed with the SEC as Exhibit 10.1 to our Quarterly Report on Form 10-Q, filed with the SEC on November 13, 2015, SEC File No. 001-36365). |
| | |
10.7* | | Amended and Restated Employment Agreement, dated December 7, 2012, between SCYNEXIS, Inc. and Charles F. Osborne, Jr. (Filed with the SEC as Exhibit 10.7 to our Registration Statement on Form S-1, filed with the SEC on February 27, 2014, SEC File No. 333-194192). |
| | |
10.8* | | Form of Stock Option Agreement and Form of Stock Option Grant Notice under the SCYNEXIS, Inc. 2014 Equity Incentive Plan (Filed with the SEC as Exhibit 99.3 to our Registration Statement on Form S-8, filed with the SEC on May 16, 2014, SEC File No. 333-196007). |
| | |
10.9* | | Amended and Restated Employment Agreement, dated December 7, 2012, between SCYNEXIS, Inc. and Yves J. Ribeill. (Filed with the SEC as Exhibit 10.9 to our Registration Statement on Form S-1, filed with the SEC on February 27, 2014, SEC File No. 333-194192). |
| | |
10.10# | | Development, License and Supply Agreement, dated August 1, 2013, between SCYNEXIS, Inc. and R-Pharm, CJSC. (Filed with the SEC as Exhibit 10.10 to our Amendment No. 1 to Registration Statement on Form S-1, filed with the SEC on March 19, 2014, SEC File No. 333-194192). |
| | |
10.11# | | License Agreement, dated August 7, 2012, as amended, between SCYNEXIS, Inc. and Dechra Ltd. (Filed with the SEC as Exhibit 10.11 to our Amendment No. 1 to Registration Statement on Form S-1, filed with the SEC on March 19, 2014, SEC File No. 333-194192). |
| | |
10.12# | | Termination and License Agreement, dated May 24, 2013, between SCYNEXIS. Inc. and Merck Sharp & Dohme Corp. (Filed with the SEC as Exhibit 10.12 to our Amendment No. 1 to Registration Statement on Form S-1, filed with the SEC on March 19, 2014, SEC File No. 333-194192). |
| | |
|
| | |
10.13# | | Agreement for the Assignment of Patents and Know How concerning Cyclosporin Derivatives, dated June 10, 2005, between SCYNEXIS, Inc. and C-CHEM AG. (Filed with the SEC as Exhibit 10.13 to our Amendment No. 1 to Registration Statement on Form S-1, filed with the SEC on March 19, 2014, SEC File No. 333-194192). |
| | |
10.14* | | Release and Settlement Agreement, dated June 30, 2015, between SCYNEXIS, Inc. and Charles F. Osborne, Jr. (Filed with the SEC as Exhibit 10.4 to our Quarterly Report on Form 10-Q, filed with the SEC on August 19, 2015, SEC File No. 001-36365). |
| | |
10.15# | | Exclusive Worldwide License Agreement, dated May 10, 2005, between SCYNEXIS, Inc. and Aventis Pharma S.A. (Filed with the SEC as Exhibit 10.15 to our Amendment No. 1 to Registration Statement on Form S-1, filed with the SEC on March 19, 2014, SEC File No. 333-194192). |
| | |
10.16 | | Amendment No. 1 to Exclusive Worldwide License Agreement, dated October 26, 2006, between SCYNEXIS, Inc. and Aventis Pharma S.A. (Filed with the SEC as Exhibit 10.16 to our Registration Statement on Form S-1, filed with the SEC on February 27, 2014, SEC File No. 333-194192). |
| | |
10.17* | | Release and Settlement Agreement, dated July 21, 2015, between SCYNEXIS, Inc. and Yves J. Ribeill, Ph.D. (Filed with the SEC as Exhibit 10.3 to our current report on Form 8-K, filed with the SEC on July 23, 2015, SEC File No. 001-36365). |
| | |
10.18* | | SCYNEXIS, Inc. 2015 Inducement Award Plan and Form of Stock Option Grant Notice and Stock Option Agreement. (Filed with the SEC as Exhibit 10.34 to our Registration Statement on Form S-1, filed with the SEC on April 9, 2015, SEC File No. 333-203314). |
| | |
10.19* | | Employment Agreement, effective November 1, 2015, between SCYNEXIS, Inc. and Eric Francois. (Filed with the SEC as Exhibit 99.1 to our current report on Form 8-K, filed with the SEC on November 2, 2015, SEC File No. 001-36365, and incorporated by reference here). |
| | |
10.20* | | Engagement Letter, dated July 7, 2015, between CMF Associates, LLC, and SCYNEXIS, Inc. (Filed with the SEC as Exhibit 10.6 to our Quarterly Report on Form 10-Q, filed with the SEC on November 13, 2015, SEC File No. 001-36365, and incorporated by reference here). |
| | |
10.21 | | Sublease Agreement, dated July 13, 2015, and effective July 22, 2015, between the Company and Optimer Pharmaceuticals, Inc. (Filed with the SEC as Exhibit 10.2 to our current report on Form 8-K, filed with the SEC on July 23, 2015, SEC File No. 001-36365). |
| | |
10.22# | | Commitment to Services Agreement, dated July 17, 2015, between SCYNEXIS, Inc. and Accuratus Lab Services, Inc. (Filed with the SEC as Exhibit 10.2 to our Quarterly Report on Form 10-Q, filed with the SEC on November 13, 2015, SEC File No. 001-36365, and incorporated by reference here). |
| | |
10.23* | | Employment Agreement, dated January 2014, between SCYNEXIS, Inc. and Carole A. Sable. (Filed with the SEC as Exhibit 10.24 to our Registration Statement on Form S-1, filed with the SEC on February 27, 2014, SEC File No. 333-194192). |
| | |
10.24* | | Employment Agreement, effective June 1, 2015, between SCYNEXIS, Inc. and David Angulo. |
| | |
10.25* | | Employment Agreement, dated February 5, 2015, between SCYNEXIS, Inc. and Dr. Marco Taglietti. (Filed with the SEC as Exhibit 10.27 to our Annual Report on Form 10-K, filed with the SEC on March 30, 2015, SEC File No. 001-36365). |
| | |
10.26 | | Patent Assignment, dated January 28, 2014, between SCYNEXIS, Inc. and Merck Sharpe & Dohme Corp. (Filed with the SEC as Exhibit 10.28 to our Registration Statement on Form S-1, filed with the SEC on February 27, 2014, SEC File No. 333-194192). |
| | |
10.27 | | Addendums to Reimbursement Agreement, dated April 9, 2010, March 17, 2014 and April 29, 2014, between SCYNEXIS, Inc. and Sanofi. (Filed with the SEC as Exhibit 10.31 to our Amendment No. 3 to Registration Statement on Form S-1, filed with the SEC on April 30, 2014, SEC File No. 333-194192). |
| | |
10.28# | | Exclusive License Agreement, dated October 29, 2014, between SCYNEXIS, Inc. and Waterstone Pharmaceutical (HK Limited). (Filed with the SEC as Exhibit 10.32 to our Annual Report on Form 10-K, filed with the SEC on March 30 2015, SEC File No. 001-36365). |
| | |
10.29# | | Amendment to Termination and License Agreement, dated December 11, 2014, between SCYNEXIS, Inc. and Merck Sharp & Dohme Corp. (Filed with the SEC as Exhibit 10.33 to our Annual Report on Form 10-K, filed with the SEC on March 30, 2015, SEC File No. 001-36365). |
| | |
23.1 | | Consent of Independent Registered Public Accounting Firm. |
| | |
31.1 | | Certification of Chief Executive Officer pursuant to Rule 13a-14(a)/15d-14(a) |
| | |
|
| | |
31.2 | | Certification of Chief Financial Officer pursuant to Rule 13(a)-14(a)/15d-14(a) |
| |
32.1 | | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18 U.S.C. Section 1350 as Adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| |
101.INS | | XBRL Instance Document |
| |
101.SCH | | XBRL Taxonomy Schema Linkbase Document |
| |
101.CAL | | XBRL Taxonomy Calculation Linkbase Document |
| |
101.DEF | | XBRL Taxonomy Definition Linkbase Document |
| |
101.LAB | | XBRL Taxonomy Labels Linkbase Document |
| | |
101.PRE | | XBRL Taxonomy Presentation Linkbase Document |
| | |
# Portions of this exhibit have been omitted pursuant to a request for confidential treatment, which portions were omitted and filed separately with the Securities and Exchange Commission. |
| | |
*Designates management contract or compensatory plan or arrangement. |